Thalassemia and Sickle Cell Anemia - Free MCQ Practice Test with solutions,
MCQ Practice Test & Solutions: Test: Thalassemia and Sickle Cell Anemia (17 Questions)
You can prepare effectively for NEET PG Medicine with this dedicated MCQ Practice Test (available with solutions) on the important topic of "Test: Thalassemia and Sickle Cell Anemia". These 17 questions have been designed by the experts with the latest curriculum of NEET PG 2026, to help you master the concept.
Test Highlights:
- - Format: Multiple Choice Questions (MCQ)
- - Duration: 30 minutes
- - Number of Questions: 17
Sign up on EduRev for free to attempt this test and track your preparation progress.

Pancytopenia may occur in the following except: (Recent Pattern 2014-15)
Detailed Solution: Question 1
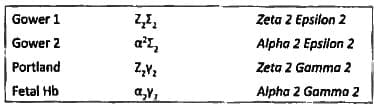
Hemoglobin with zeta 2 and gamma 2 chains are seen in which of the following: (Recent Pattern 2014-15)
Detailed Solution: Question 2

The most appropriate drug used for chelation therapy in beta thalassemia major is: (Recent Pattern 2014-15)
Detailed Solution: Question 3
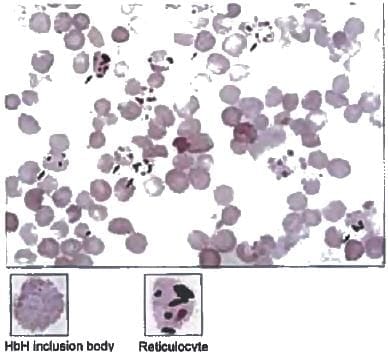
HbH is characterized by: (Recent Pattern 2014-15)
Detailed Solution: Question 4

Which of the following is not seen in a chronic case of Sickle cell anemia: (AI 1996)
Detailed Solution: Question 5
All are true for sickle cell anemia, except (AIIMS May 94)
Detailed Solution: Question 6
Golf ball inclusion bodies in RBCs are seen in? (Recent Question 2016-17)
Detailed Solution: Question 7
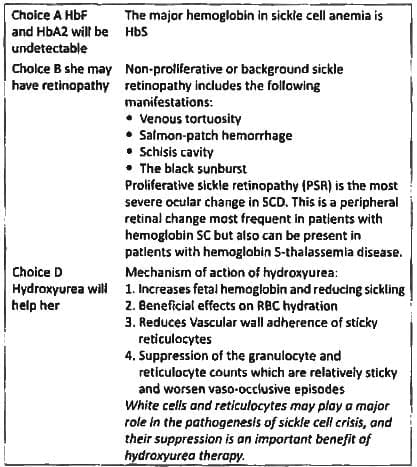
A 25-year-old lady came with anemia jaundice and recurrent joint pains. All of the following are true except: (APPG 2015)
Detailed Solution: Question 8
The abnormality in X-ray skull shown can be seen in the following conditions except: (APPG 2015)

Detailed Solution: Question 9
Persistent priapism is due to: (Recent Pattern 2014-15)
Detailed Solution: Question 10
Regarding to Thalassemia minor the following is incorrect: (Recent Pattern 2014-15)
Detailed Solution: Question 11
Basic defect in HbS is: (Recent Pattern 2014-15)
Detailed Solution: Question 12
In Beta thalassemia, the most common gene mutation is: (Recent Pattern 2014-15)
Detailed Solution: Question 13

Fetal hemoglobin achieves adult values by: (Recent Pattern 2014-15)
Detailed Solution: Question 14
Sickle cell anemia is usually associated with all, except: (Recent Pattern 2014-15)
Detailed Solution: Question 15
X-ray skull characteristically shows “Hair standing on end” appearance in one of the following disease: (Recent Pattern 2014-15)
Detailed Solution: Question 16
Auto-splenectomy is associated with: (Recent Pattern 2014-15)
Detailed Solution: Question 17
52 docs|64 tests |