Organic Reactions With Mechanism and Applications (Part -5) | Organic Chemistry PDF Download
⇒ Dienone - Phenol Rearrangement:
When 4, 4-dialkyl cyclohexadienone is treated with acid, it is converted to phenol with migration of one of the alkyl groups to the adjacent carbon. This is known as dienone-phenol rearrangement. The dienone is dissolved in acetic anhydride and treated with catalytic amount of sulphuric acid. The product on hydrolysis gives the phenol.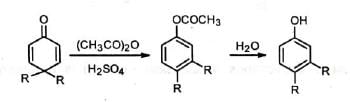
Mechanism:
On protonation of the oxygen, a carbocation is generated which is stabilised by delocalization of the positive charge.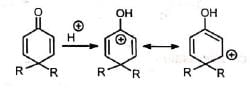 In one of the canonical structures, the positive charge is on a carbon adjacent to a highly substituted carbon. Hence, a carbocation rearrangement occurs. Subsequent loss of a proton gives the 3, 4-disubstituted phenol.
In one of the canonical structures, the positive charge is on a carbon adjacent to a highly substituted carbon. Hence, a carbocation rearrangement occurs. Subsequent loss of a proton gives the 3, 4-disubstituted phenol.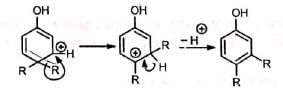 The ease of dienone-phenol rearrangement is due to the creation of a stable aromatic system.
The ease of dienone-phenol rearrangement is due to the creation of a stable aromatic system.
When one of the alkyl group forms a part of the cyclic system, either the alkyl group or the ring methylene group may migrate.
(i) Alkyl migration: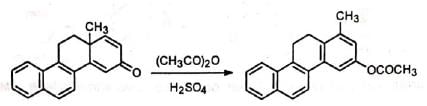 (ii) Migration of ring methylene group:
(ii) Migration of ring methylene group: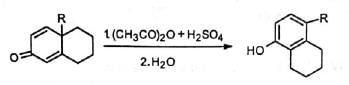 The course of the reaction depends on the structural or electronic factors and on the conditions of reaction.
The course of the reaction depends on the structural or electronic factors and on the conditions of reaction.
A reverse rearrangement, i.e., phenol-dienone rearrangement has been observed during the electrophilic substitution in phenols in some cases.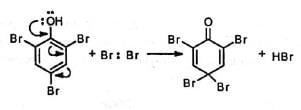
Applications:
The rearrangement has useful applications. A classical example is the rearrangement of santonin to desmotropo santonin.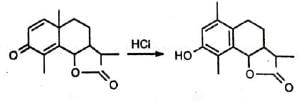 The anthrasteroid rearrangement is closely related.
The anthrasteroid rearrangement is closely related.
⇒ Benzilic Acid Rearrangement:
The addition of a strong base to a carbonyl group results in the formation of an anion. The reversal of the anionic charge may cause expulsion of the attached group, X, e.g.,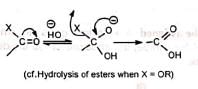 However, in a 1,2-diketone the group X may migrate to the adjacent electron-deficient carbonyl carbon forming α-hydroxy acid.
However, in a 1,2-diketone the group X may migrate to the adjacent electron-deficient carbonyl carbon forming α-hydroxy acid.
Thus, benzil on treatment with a strong base forms benzilic acid (salt), hence the name benzilic acid rearrangement.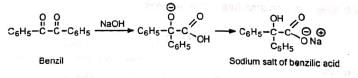 Barium and thallous hydroxides are more effective than sodium or potassium hydroxides. Alkoxide ions (methoxide, t-butoxide, etc.) in place of hydroxide ion give the corresponding esters.
Barium and thallous hydroxides are more effective than sodium or potassium hydroxides. Alkoxide ions (methoxide, t-butoxide, etc.) in place of hydroxide ion give the corresponding esters.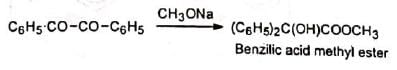 Phenoxide ions are too weak a nucleophile to attack. Besides aromatic 1, 2-diketones, aliphatic and heterocyclic diketones as also o-quinones undergo this rearrangement.
Phenoxide ions are too weak a nucleophile to attack. Besides aromatic 1, 2-diketones, aliphatic and heterocyclic diketones as also o-quinones undergo this rearrangement.
Mechanism:
It has been seen that the rate of reaction is proportional to the concentrations of benzil and the hydroxide ion, i.e., rate 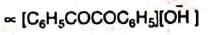
It has also been found that when the reaction is carried out in the presence of H218O, benzil exchanges 18O faster than it rearranges.
On the basis of the above observations, it has been suggested that a fast reversible nucleophilic attack occurs at the carbonyl carbon in the first step. The second step is the rate-determining step in which the migration occurs. Finally, a rapid proton transfer completes the process.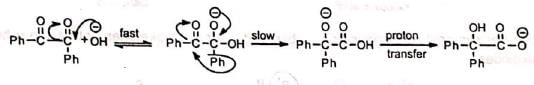 The rearrangement is analogous to intramolecular Cannizzaro reaction of glyoxal.
The rearrangement is analogous to intramolecular Cannizzaro reaction of glyoxal.
The carbonyl group which is attached to the less electron releasing of the two aryl groups is relatively more positively charged and, hence, is attacked by OH. Consequently, the less electron-donating aryl group migrates to the other carbonyl group.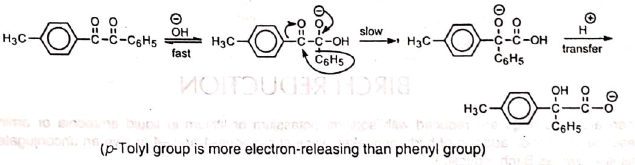
Applications:
The reaction is a general one and can take place with aromatic, heterocyclic, alicyclic, and aliphatic 1 , 2-diketones as also 1 , 2-quinones.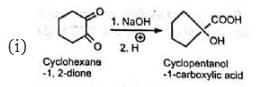
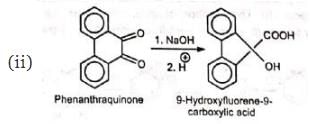
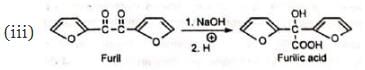
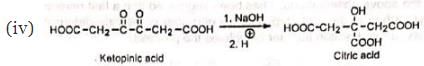
(v) Similar benzilic acid rearrangement is observed when α-haloketones not having α-hydrogens are treated with alkoxides.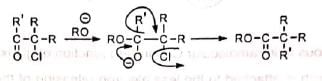 This reaction is known as semibenzilic rearrangement.
This reaction is known as semibenzilic rearrangement.
⇒ Baeyer - Villiger Rearrangement:
Baeyer-Villiger rearrangement is an example of the migration of a group from carbon to electron-deficient oxygen.
The reaction involves the oxidation of ketones to esters by the treatment with peracids such as peracetic acid, perbenzoic acid, pertrifluoroacetic acid, permonosulphuric acid. etc.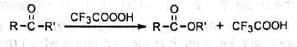 Cyclic ketones are converted to lactones with ring expansion.
Cyclic ketones are converted to lactones with ring expansion.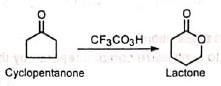 The overall reaction is an insertion of oxygen atom between the carbonyl group and the adjacent carbon in ketone. Organic solvents which are inert under the conditions of reaction may be used. The choice of solvent depends upon the solubility of the reactants. Commonly used solvents are glacial acetic acid and chloroform.
The overall reaction is an insertion of oxygen atom between the carbonyl group and the adjacent carbon in ketone. Organic solvents which are inert under the conditions of reaction may be used. The choice of solvent depends upon the solubility of the reactants. Commonly used solvents are glacial acetic acid and chloroform.
Mechanism:
Nucleophilic attack of the peracid on the protonated ketone gives an intermediate peroxide (i). The peroxide then undergoes loss of carboxylate anion and migration of a group from carbon to electron deficient oxygen to yield the protonated ester
(ii). Finally the loss of proton gives the ester.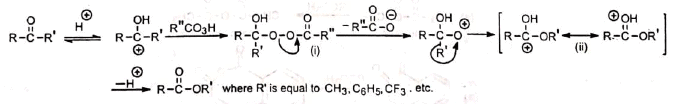 The reaction is catalysed by acids. Electron-releasing groups in the ketone and electron-withdrawing groups in peracids promote the reaction rate. Pertrifluoroacetic acid is very effective because trifluoroacetate ion is a good leaving group.
The reaction is catalysed by acids. Electron-releasing groups in the ketone and electron-withdrawing groups in peracids promote the reaction rate. Pertrifluoroacetic acid is very effective because trifluoroacetate ion is a good leaving group.
The mechanism is supported by the fact that the labelled oxygen atom of the ketone is entirely present in the carbonyl oxygen of the ester.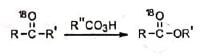 The loss of carboxylate anion and the migration of the group may be concerted. Syrkin has suggested that the peroxide transforms into products by a cyclic mechanism, which shows that the last three steps may be concerted.
The loss of carboxylate anion and the migration of the group may be concerted. Syrkin has suggested that the peroxide transforms into products by a cyclic mechanism, which shows that the last three steps may be concerted.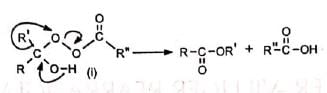 The migrating group retains its configuration as in other concerted reactions. For acylic compounds the migrating group, R' must be 2°, 3° or vinylic. However, migration of 1° alkyl group may be brought about by using CF3CO3H or BF3- H2O2 as reagent.
The migrating group retains its configuration as in other concerted reactions. For acylic compounds the migrating group, R' must be 2°, 3° or vinylic. However, migration of 1° alkyl group may be brought about by using CF3CO3H or BF3- H2O2 as reagent.
Baeyer-Villiger oxidation can be brought about with H2O2 and base also in some cases.
In unsymmetrical ketones, that group migrates which is more electron-releasing. Thus, the migratory aptitude of alkyl groups is in the order 3° > 2° > 1° > CH3. Electron-releasing substituents in the aryl group facilitate migration. The migratory order of aryl groups is p-anisyl > p-tolyl > phenyl > p-chlorophenyl > p-nitrophenyl, etc. In case of alkyl aryl ketones, it is the aryl group which migrates (except in case of t-butyl group).
Applications:
The reaction has valuable synthetic applications.
1. Esters: Esters which are difficult to synthesize can be prepared by this method.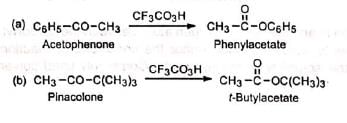 2. Anhydrides: When 1, 2-diketones or o-quinones are subjected to Baeyer-Villiger rearrangement, anhydrides are produced.
2. Anhydrides: When 1, 2-diketones or o-quinones are subjected to Baeyer-Villiger rearrangement, anhydrides are produced.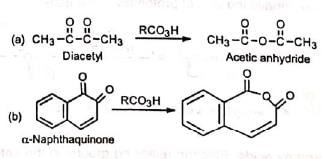
The products can be converted to various types of compounds.
3. Lactones: Cyclic ketones are converted to lactones with ring expansion.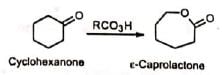 Long-chain hydroxyesters can be prepared from large ring-size ketones.
Long-chain hydroxyesters can be prepared from large ring-size ketones.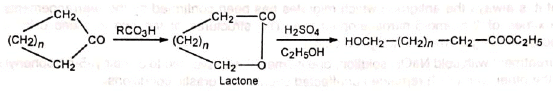 With some condensed cyclic ketones, two lactones in varying proportions are formed. For example, camphor gives two lactones (I) and (II).
With some condensed cyclic ketones, two lactones in varying proportions are formed. For example, camphor gives two lactones (I) and (II).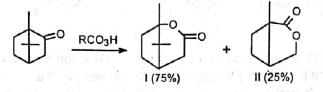 Lactone (I) is the normal product formed by the migration of the tertiary bridgehead carbon while lactone (II) has been formed by the migration of the methylene group. The reason for the formation of two lactones in different proportions is steric factor.
Lactone (I) is the normal product formed by the migration of the tertiary bridgehead carbon while lactone (II) has been formed by the migration of the methylene group. The reason for the formation of two lactones in different proportions is steric factor.
4. Elucidation of structure: The ester obtained as a result of the rearrangement may be hydrolysed to acid and alcohol from which the structure of the substrate can be determined.
The reaction is not successful with aldehydes Aliphatic aldehydes are oxidized to acids by the migration of the hydrogen.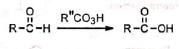 A few aromatic aldehydes have been converted to formates by the migration of the aryl group.
A few aromatic aldehydes have been converted to formates by the migration of the aryl group.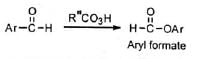
⇒ Diels - Alder Reaction:
Diels-Alder reaction involves the 1, 4-addition of an alkene to a conjugated diene to form an adduct of six-membered ring. The double bond compound is called the dienophile. The reaction is initiated thermally or by Lewis acid catalyst with or without the use of solvents.
Ethylene and simple olefins give poor yields even at high temperature.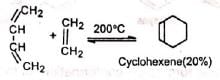 Electron-withdrawing substituents in the dienophile, such as >C=O, -CHO, -COOR, -CN , -NO2, etc., promote the reaction.
Electron-withdrawing substituents in the dienophile, such as >C=O, -CHO, -COOR, -CN , -NO2, etc., promote the reaction.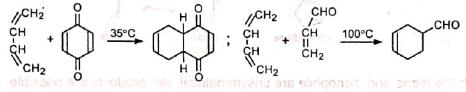 The reaction rate is also accelerated by the presence of electron-releasing groups in the dienes. Thus, the reaction between 2-methyl-1, 3-butadiene (isoprene) and acraldehyde is faster than that between 1, 3-butadiene and acraldehyde.
The reaction rate is also accelerated by the presence of electron-releasing groups in the dienes. Thus, the reaction between 2-methyl-1, 3-butadiene (isoprene) and acraldehyde is faster than that between 1, 3-butadiene and acraldehyde.
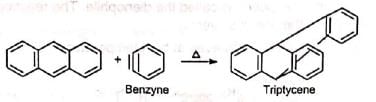
Triple bond compounds, allenes and benzyne may also function as dienophiles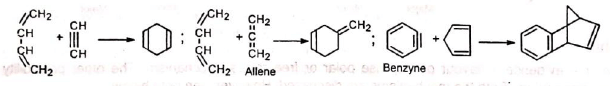 Since the reaction involves uniting of π orbitals, all carbon skeleton is not necessary in the dienophile, e.g.,
Since the reaction involves uniting of π orbitals, all carbon skeleton is not necessary in the dienophile, e.g.,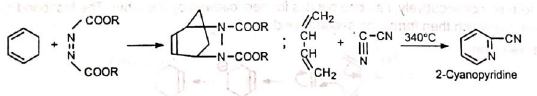 Besides acylic hydrocarbons, dienes may be alicyclic hydrocarbons, in which the conjugated double bonds may be wholly or partly inside the ring, some heterocyclic and some aromatic hydrocarbons.
Besides acylic hydrocarbons, dienes may be alicyclic hydrocarbons, in which the conjugated double bonds may be wholly or partly inside the ring, some heterocyclic and some aromatic hydrocarbons.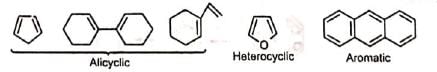 Benzene, naphthalene and phenanthreno are quite unreactive but anthracene responds readily.
Benzene, naphthalene and phenanthreno are quite unreactive but anthracene responds readily.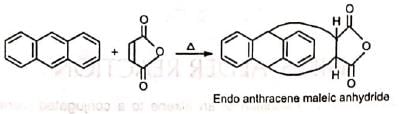
 The reaction occurs with the cisoid form (s-cis conformation) of the diene. Cyclic dienes are naturally in cisoid form and so react more rapidly than the acyclic dienes which normally exist in the more stable transoid forms (s-trans conformation).
The reaction occurs with the cisoid form (s-cis conformation) of the diene. Cyclic dienes are naturally in cisoid form and so react more rapidly than the acyclic dienes which normally exist in the more stable transoid forms (s-trans conformation).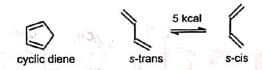 When both the diene and dienophile are unsymmetrical, two products are possible. However, the 1, 2- and 1, 4-products predominate.
When both the diene and dienophile are unsymmetrical, two products are possible. However, the 1, 2- and 1, 4-products predominate.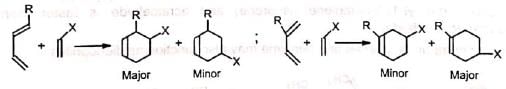
Mechanism:
There is little evidence in favour of stepwise polar or free radical mechanism. The other possibility is a concerted mechanism. Both the mechanisms are discussed. However, see note below.
(i) Stepwise polar and free radical mechanism: The mechanism envisages that the bonds between the reactants are formed consecutively, i.e., one bond is formed followed by the other. The first bond gives a diion (a) or a diradical (b) which then forms the second bond.
(a) Polar reaction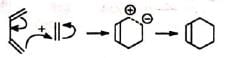 (b) Free radical reaction
(b) Free radical reaction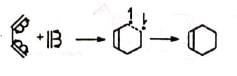 The mechanism assumes that the second bond is formed more rapidly than the rotation about the C-C bond, as otherwise a mixture of cis and trans products (nonstereospecific) would be obtained with substituted reactants, which is contrary to observation. The reaction is strictly stereospecific. The orientation of the groups (i.e., cis or trans) in the reactant remain unaltered in the product. Hence the mechanism is not favourable.
The mechanism assumes that the second bond is formed more rapidly than the rotation about the C-C bond, as otherwise a mixture of cis and trans products (nonstereospecific) would be obtained with substituted reactants, which is contrary to observation. The reaction is strictly stereospecific. The orientation of the groups (i.e., cis or trans) in the reactant remain unaltered in the product. Hence the mechanism is not favourable.
(ii) Concerted mechanism: The mechanism visualises a process in which there is simultaneous (not consecutive) breaking and making of bonds through a six-centred transition state with no intermediate. It is a 4π + 2π cycloaddition and stereospecifically cis with respect to both reactants.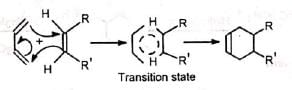 The addition of the dienophile is always cis so that the groups cis in the olefin remain cis in the adduct. The addition is therefore stereospecific.
The addition of the dienophile is always cis so that the groups cis in the olefin remain cis in the adduct. The addition is therefore stereospecific.
There are two possible modes of addition, (a) exo and (b) endo. When the greater part of the dienophile is under the diene ring in the adduct, it is called endo and in the reverse case, it is called exo. The endo adduct is more stable.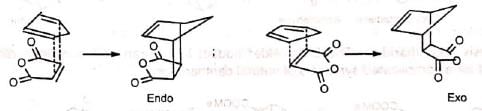 This is because the cyclic transition state has further stabilization through secondary 7i-orbital overlaps for greater accumulation of double bonds in the endo adduct.
This is because the cyclic transition state has further stabilization through secondary 7i-orbital overlaps for greater accumulation of double bonds in the endo adduct.
Applications:
The reaction has proved of great value. Due to its high stereospecific nature, the reaction has been used to synthesize many natural products which otherwise would be difficult to prepare. The reaction can be used as a diagnostic test for conjugation in a system. Since the reaction is stereospecific, the configurations of the reactants can be determined by studying the adduct. The reaction has also been used to trap benzyne intermediate. Some reactions are given to illustrate its use.
1. Determination of configuration: Maleic and fumaric acids can be identified from the products.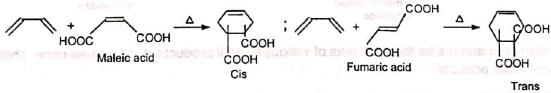 2. Synthesis of quinones and polynuclear hydrocarbons:
2. Synthesis of quinones and polynuclear hydrocarbons:
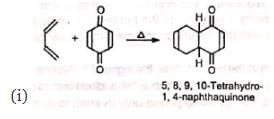
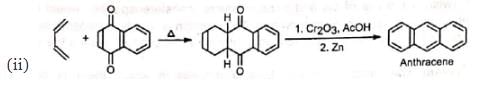
3. Synthesis of α-terpineol: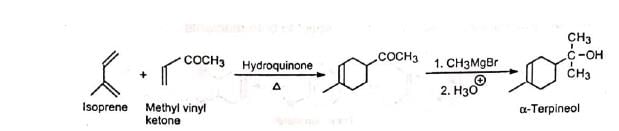 4. In the synthesis of camphene: The intermediate required in the synthesis of camphene is prepared by Diels-Alder reaction.
4. In the synthesis of camphene: The intermediate required in the synthesis of camphene is prepared by Diels-Alder reaction.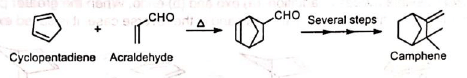 5. In the synthesis of cantharidine: The Diels-Alder adduct from furan and acetylene dicarboxylate is the starting product for a complicated synthesis of natural cantharidine.
5. In the synthesis of cantharidine: The Diels-Alder adduct from furan and acetylene dicarboxylate is the starting product for a complicated synthesis of natural cantharidine.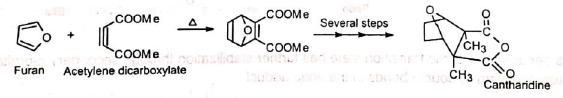 The Diels-Alder adduct obtained from furan and dimethylmaleic anhydride on hydrogenation gives cantharidine which is not the same as the natural product.
The Diels-Alder adduct obtained from furan and dimethylmaleic anhydride on hydrogenation gives cantharidine which is not the same as the natural product.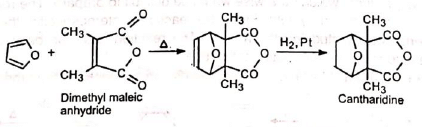 The starting materials for the synthesis of various natural products, such as reserpine, cholesterol, etc., are the Diels-Alder adducts.
The starting materials for the synthesis of various natural products, such as reserpine, cholesterol, etc., are the Diels-Alder adducts.
Note: The concerted cycloaddition is best explained from the principle of Conservation of Orbital Symmetry put forward by Woodward and Hoffmann. The principle states that cycloaddition is allowed when the symmetry properties of the highest occupied molecular orbital (HOMO) of one reactant correlates with the lowest unoccupied molecular orbital (LUMO) of the other and vice versa.
It is seen that in the reactants in the ground state, the signs of 1 , 4-lobes of lumo for 1, 3-butadiene correlates with those of homo of ethylene, and hence the addition occurs smoothly on heating.
(Orbitals of the same signs, i.e., of same phase only overlap to form bonds.)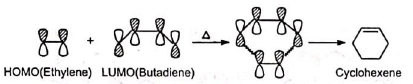 Similar correlation is observed between butadiene homo and ethylene lumo.
Similar correlation is observed between butadiene homo and ethylene lumo.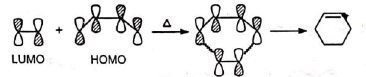 The stereospecificity (i.e., cis-addition) is thus explained.
The stereospecificity (i.e., cis-addition) is thus explained.
|
35 videos|92 docs|46 tests
|
FAQs on Organic Reactions With Mechanism and Applications (Part -5) - Organic Chemistry
| 1. What are some common types of organic reactions? |  |
| 2. How do substitution reactions occur in organic chemistry? | |
| 3. What are the applications of organic reactions? | |
| 4. How do oxidation-reduction reactions occur in organic chemistry? | |
| 5. What is the mechanism behind elimination reactions in organic chemistry? | |

|
4.98/5 Rating |

|
Dec 23, 2024 Last updated |

|
Explore Courses for Chemistry exam
|
|
study material
,Previous Year Questions with Solutions
,Summary
,past year papers
,Organic Reactions With Mechanism and Applications (Part -5) | Organic Chemistry
,Organic Reactions With Mechanism and Applications (Part -5) | Organic Chemistry
,practice quizzes
,shortcuts and tricks
,Extra Questions
,MCQs
,Free
,Objective type Questions
,Organic Reactions With Mechanism and Applications (Part -5) | Organic Chemistry
,Semester Notes
,Important questions
,Viva Questions
,ppt
,mock tests for examination
,Exam
,video lectures
,Sample Paper
;






Organic Reactions With Mechanism and Applications (Part -5) Free PDF Download
Importance of Organic Reactions With Mechanism and Applications (Part -5)
Organic Reactions With Mechanism and Applications (Part -5) Notes
Organic Reactions With Mechanism and Applications (Part -5) Chemistry Questions
Study Organic Reactions With Mechanism and Applications (Part -5) on the App
|
© EduRev
|
Education Revolution
|



|




