Organic Reactions With Mechanism and Applications (Part -4) | Organic Chemistry PDF Download
⇒Pinacol - Pinacolone Rearrangement:
The acid-catalysed rearrangement of vie diols (1, 2-diols) to ketones or aldehydes with elimination of water is known as pinacol or pinacol-pinacolone rearrangement. The name was given from the classical example of the conversion of pinacol to pinacolone.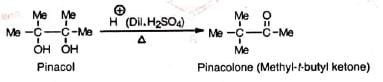
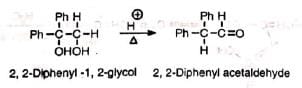 Elimination of water without rearrangement— the normal reaction of alcohols— may be achieved under drastic conditions (Al2 O3 , 450°C).
Elimination of water without rearrangement— the normal reaction of alcohols— may be achieved under drastic conditions (Al2 O3 , 450°C).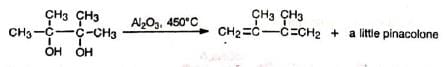 The rearrangement has been successfully carried out with various polysubstituted glycols.
The rearrangement has been successfully carried out with various polysubstituted glycols.
Mechanism:
The reaction involves the general characteristics of carbocation rearrangement in which the driving force is the stabilization of the resulting carbocation.
The reaction starts with the protonation of the hydroxyl group followed by elimination of water and formation of carbocation (I). The carbocation is then stabilized by Whitmore 1, 2-shift. Finally, elimination of a proton from the stable carbocation (II) gives the carbonyl compound.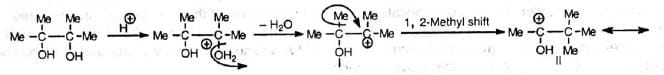
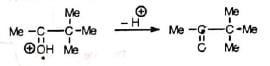 The carbocation (I), though tertiary, prefers to form (II) for its resonance stability.
The carbocation (I), though tertiary, prefers to form (II) for its resonance stability.
The mechanism is supported by the fact that any carbocation in which the positive charge is on the carbon adjacent to the one bearing the hydroxyl group 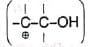 also undergoes similar rearrangement. Thus,
also undergoes similar rearrangement. Thus,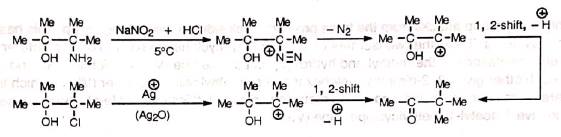 The loss of water and migration of the alkyl group may be very rapid or simultaneous. Probably the migrating group does not become completely free before it is partially bonded (III) to the adjacent positively charged carbon, i.e., a type of intramolecular rearrangement is suggested.
The loss of water and migration of the alkyl group may be very rapid or simultaneous. Probably the migrating group does not become completely free before it is partially bonded (III) to the adjacent positively charged carbon, i.e., a type of intramolecular rearrangement is suggested.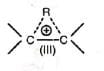 Evidence in favour of this are (a) the migrating group retains its configuration, if chiral and (b) no cross-over products are obtained when a mixture of two nearly similar 1,2-diols is treated with acid.
Evidence in favour of this are (a) the migrating group retains its configuration, if chiral and (b) no cross-over products are obtained when a mixture of two nearly similar 1,2-diols is treated with acid.
Migratory aptitude Migration order in general is H > aryl > alkyl. As the migrating group migrates with its electron pair, the more nucleophilic group might be expected to migrate. Thus, the order of migration amongst the aryl groups is p-anisyl > p-tolyl > phenyl > p-chlorophenyl, etc.
As the migrating group migrates with its electron pair, the more nucleophilic group might be expected to migrate. Thus, the order of migration amongst the aryl groups is p-anisyl > p-tolyl > phenyl > p-chlorophenyl, etc.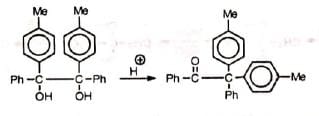 Obviously, electron-withdrawing groups will retard the migration. Tho migratory aptitude amongst tho alkyl groups is Me3C > Me2CH > Me. However, the stability of tho Initially formed carbocation may offset tho migratory aptitude order. The initial carbocation is formed by tho loss of that hydroxyl group which results In the formation of the most stable carbocation. Thus, In tho compound 1, 1-dimethyl-2, 2-diphenyl glycol, the resonance-stabilized carbocation (IV) is formed Instead of (V) and so it Is tho methyl group and not the phenyl group which migrates, contrary to the above sequence
Obviously, electron-withdrawing groups will retard the migration. Tho migratory aptitude amongst tho alkyl groups is Me3C > Me2CH > Me. However, the stability of tho Initially formed carbocation may offset tho migratory aptitude order. The initial carbocation is formed by tho loss of that hydroxyl group which results In the formation of the most stable carbocation. Thus, In tho compound 1, 1-dimethyl-2, 2-diphenyl glycol, the resonance-stabilized carbocation (IV) is formed Instead of (V) and so it Is tho methyl group and not the phenyl group which migrates, contrary to the above sequence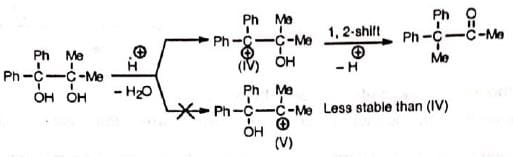 Steric hindrance may affect the rate of migration— p-anisyl group migrates 1000 times faster than o-anisyl group.
Steric hindrance may affect the rate of migration— p-anisyl group migrates 1000 times faster than o-anisyl group.
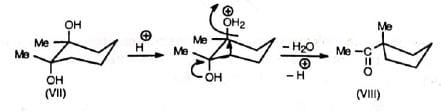
The migrating group attacks from the trans position (back side) to the leaving group. This has important effect in cyclic systems. Thus, the two isomers of 1, 2-dimethyl-cyclohexane-1,2-diol give different products due to different orientations of the methyl and hydroxyl groups. The one (VI) in which the Me and OH groups are trans to each other gives 2, 2-dimethylcyclohexanone by methyl shift. The other (VII) in which the Me and OH groups are c/s to each other undergoes ring methylene group shift Instead of Me-shift with consequent ring contraction to give 1-acetyl-1-methylcyclopentane (VIII).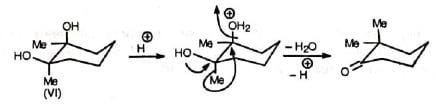
 The rearrangement has interesting applications in synthesis.
The rearrangement has interesting applications in synthesis.
Applications:
1. Synthesis of carbonyl compounds from alkenes Isobutyraldehyde may be prepared on a large scale from isobutylene.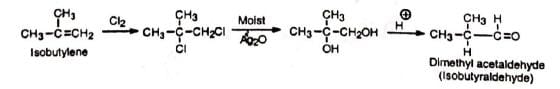 2. Ring expansion of cyclic ketones Cyclohexanone can be converted to cycloheptanone in good yield
2. Ring expansion of cyclic ketones Cyclohexanone can be converted to cycloheptanone in good yield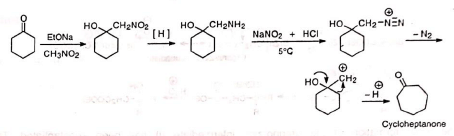 3. Ketones from cyclic diols Pinacol rearrangement has been employed to prepare ketones which are otherwise inaccessible or are very difficult to synthesize.
3. Ketones from cyclic diols Pinacol rearrangement has been employed to prepare ketones which are otherwise inaccessible or are very difficult to synthesize.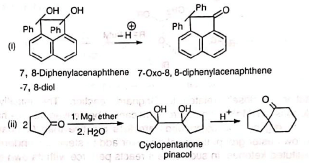
⇒ Favorskii Rearrangement:
The transformation of α-haloketones to esters with rearranged carbon skeleton by the treatment with alkoxide ions is called Favorskii rearrangement. Alkali hydroxides or amines in place of metal alkoxides give acids or amides respectively.
Cyclic α-haloketones give esters with ring contraction.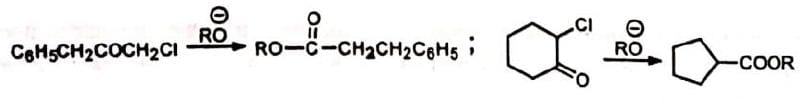
Mechanism:
The mechanism of the rearrangement has been the subject of much investigation.
It was observed that both the isomeric ketones. (I) and (II) gave β-phenylpropionic acid on treatment with hydroxide ions. This observation indicates that the chlorine is not being directly replaced by the incoming group from the other side of the carbonyl group, as otherwise (II) would give C6H5CH(CH3)COOH which is not obtained. Further, it was observed that a cyclic ketone (2-chlorocyclohexanone) with labelled carbon bearing the chlorine atom, on treatment with alkoxide ion gave a product in which equal amounts of 14C were present at the α-carbon and at the β-carbon. This suggests a symmetrical cyclopropanone intermediate which opens up with equal ease on either side of the carbonyl group.
This observation indicates that the chlorine is not being directly replaced by the incoming group from the other side of the carbonyl group, as otherwise (II) would give C6H5CH(CH3)COOH which is not obtained. Further, it was observed that a cyclic ketone (2-chlorocyclohexanone) with labelled carbon bearing the chlorine atom, on treatment with alkoxide ion gave a product in which equal amounts of 14C were present at the α-carbon and at the β-carbon. This suggests a symmetrical cyclopropanone intermediate which opens up with equal ease on either side of the carbonyl group.
On the basis of the above observations, the following mechanism has been suggested.
The base abstracts an a-hydrogen to produce a carbanion. Intramolecular nucleophilic attack on the carbon bearing the chlorine displaces the chlorine atom with the formation of a transient symmetrical cyclopropanone ring. Subsequent attack of the alkoxide ion on the carbonyl carbon opens the ring with equal ease on either side of the carbonyl carbon so that the product contains 50% of 14C at the a-position and 50% at the β-position.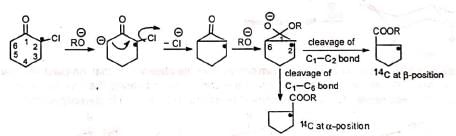 In case of unsymmetrical ketones, the unsymmetrical cyclopropanone ring which is formed, opens up to give the most stable carbanion. Thus, the two isomeric ketones (I) and (II) give the same cyclic intermediate (III) which may open on either side of the carbonyl group to give two carbanions (IV) and (V).
In case of unsymmetrical ketones, the unsymmetrical cyclopropanone ring which is formed, opens up to give the most stable carbanion. Thus, the two isomeric ketones (I) and (II) give the same cyclic intermediate (III) which may open on either side of the carbonyl group to give two carbanions (IV) and (V).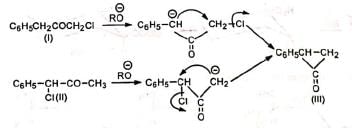
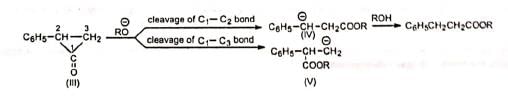 The carbanion (IV) being resonance-stabilized is preferentially formed so that the product is C6H5CH2CH2COOR. Hence, both (I) and (II) give the same product.
The carbanion (IV) being resonance-stabilized is preferentially formed so that the product is C6H5CH2CH2COOR. Hence, both (I) and (II) give the same product.
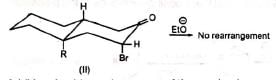
Although the cyclopropanone intermediate has not been isolated, it has been trapped to give an adduct with furan in one case at least.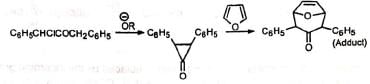 The formation of cyclopropanone intermediate probably proceeds via an intramolecular 1,3-elimination involving a backside attack of the carbanion. Thus, in nonpolar media the cyclic α-haloketone (I) with equatorial halogen underwent rearrangement but (II) with axial halogen did not, since the backside attack is restricted. (Stork and Borowitz)
The formation of cyclopropanone intermediate probably proceeds via an intramolecular 1,3-elimination involving a backside attack of the carbanion. Thus, in nonpolar media the cyclic α-haloketone (I) with equatorial halogen underwent rearrangement but (II) with axial halogen did not, since the backside attack is restricted. (Stork and Borowitz)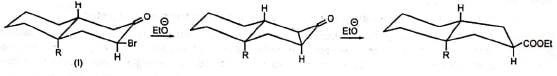
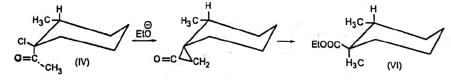
 Additional evidence in support of the mechanism comes from the observation that the diastereomers (III) and (IV) gave (V) and (VI) respectively indicating that the carbon atom bearing the chlorine atom underwent inversion as is the requirement for SN2 displacement reactions. The reaction is stereospecific.
Additional evidence in support of the mechanism comes from the observation that the diastereomers (III) and (IV) gave (V) and (VI) respectively indicating that the carbon atom bearing the chlorine atom underwent inversion as is the requirement for SN2 displacement reactions. The reaction is stereospecific.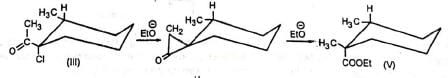
 For simplicity, this reaction may be represented as given below.
For simplicity, this reaction may be represented as given below.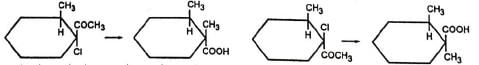 Ketones not having α-hydrogen also undergo similar rearrangement in some cases. The mechanism for this is, in part, similar to benzilic acid rearrangement and is called semibenzilic mechanism.
Ketones not having α-hydrogen also undergo similar rearrangement in some cases. The mechanism for this is, in part, similar to benzilic acid rearrangement and is called semibenzilic mechanism.
Stereospecific reaction-When a given stereomer gives one product while the other stereomer gives the opposite product, the reaction is called stereospecific.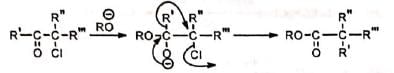 Even when there is a suitably placed ct-hydrogen, the rearrangements may follow semibenzilic pattern in some compounds.
Even when there is a suitably placed ct-hydrogen, the rearrangements may follow semibenzilic pattern in some compounds.
Thus, 2-bromocyclobutanone undergoes Favorskii rearrangement when treated with water as the base. When treated with D20 , no deuterium is incorporated in the ring.
Cyclopropanone intermediate mechanism suggests incorporation of deuterium in the ring.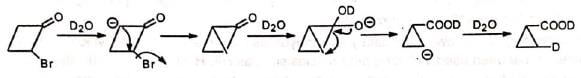 However, semibenzilic mechanism can explain the formation of the actual product without deuterium in the ring.
However, semibenzilic mechanism can explain the formation of the actual product without deuterium in the ring.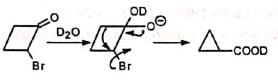 Probably, the strain in the bicyclobutanone ring restricts the operation of cyclopropanone mechanism.
Probably, the strain in the bicyclobutanone ring restricts the operation of cyclopropanone mechanism.
Favorskii rearrangement of α1α (gem) and αα' (vic) dihaloketones produce α, β-unsaturated esters.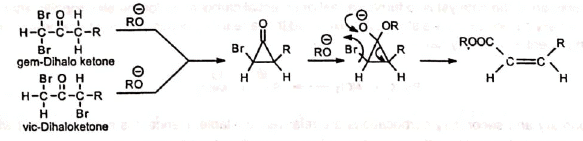 The reaction is stereoselective as it gives cis olefin.
The reaction is stereoselective as it gives cis olefin.
|
35 videos|92 docs|46 tests
|
FAQs on Organic Reactions With Mechanism and Applications (Part -4) - Organic Chemistry
| 1. What is an organic reaction? |  |
| 2. What is a mechanism in organic reactions? | |
| 3. What are some applications of organic reactions? | |
| 4. How are mechanisms determined in organic reactions? | |
| 5. What are the factors that can influence the outcome of organic reactions? | |

|
4.98/5 Rating |

|
Dec 23, 2024 Last updated |

|
Explore Courses for Chemistry exam
|
|
study material
,Viva Questions
,MCQs
,practice quizzes
,Organic Reactions With Mechanism and Applications (Part -4) | Organic Chemistry
,Organic Reactions With Mechanism and Applications (Part -4) | Organic Chemistry
,Previous Year Questions with Solutions
,video lectures
,shortcuts and tricks
,Extra Questions
,ppt
,Exam
,Free
,Organic Reactions With Mechanism and Applications (Part -4) | Organic Chemistry
,Semester Notes
,Important questions
,Summary
,Objective type Questions
,mock tests for examination
,Sample Paper
,past year papers
;






Organic Reactions With Mechanism and Applications (Part -4) Free PDF Download
Importance of Organic Reactions With Mechanism and Applications (Part -4)
Organic Reactions With Mechanism and Applications (Part -4) Notes
Organic Reactions With Mechanism and Applications (Part -4) Chemistry Questions
Study Organic Reactions With Mechanism and Applications (Part -4) on the App
|
© EduRev
|
Education Revolution
|



|




