Diels-Alder reaction involves the 1, 4-addition of an alkene to a conjugated diene to form an adduct of six-membered ring. The double bond compound is called the dienophile. The reaction is initiated thermally or by Lewis acid catalyst with or without the use of solvents.
Ethylene and simple olefins give poor yields even at high temperature.
 Electron-withdrawing substituents in the dienophile, such as >C=O, -CHO, -COOR, -CN , -NO2, etc., promote the reaction.
Electron-withdrawing substituents in the dienophile, such as >C=O, -CHO, -COOR, -CN , -NO2, etc., promote the reaction.
 The reaction rate is also accelerated by the presence of electron-releasing groups in the dienes. Thus, the reaction between 2-methyl-1, 3-butadiene (isoprene) and acraldehyde is faster than that between 1, 3-butadiene and acraldehyde.
The reaction rate is also accelerated by the presence of electron-releasing groups in the dienes. Thus, the reaction between 2-methyl-1, 3-butadiene (isoprene) and acraldehyde is faster than that between 1, 3-butadiene and acraldehyde.
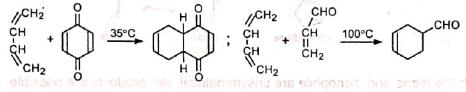
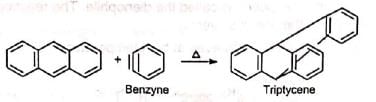
Triple bond compounds, allenes and benzyne may also function as dienophiles
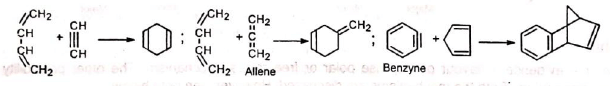 Since the reaction involves uniting of π orbitals, all carbon skeleton is not necessary in the dienophile, e.g.,
Since the reaction involves uniting of π orbitals, all carbon skeleton is not necessary in the dienophile, e.g.,
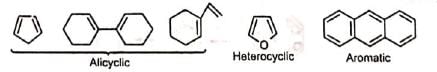
 Besides acylic hydrocarbons, dienes may be alicyclic hydrocarbons, in which the conjugated double bonds may be wholly or partly inside the ring, some heterocyclic and some aromatic hydrocarbons.
Besides acylic hydrocarbons, dienes may be alicyclic hydrocarbons, in which the conjugated double bonds may be wholly or partly inside the ring, some heterocyclic and some aromatic hydrocarbons.
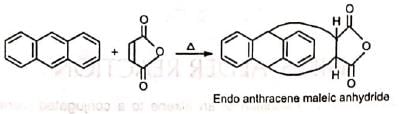
 Benzene, naphthalene and phenanthreno are quite unreactive but anthracene responds readily.
Benzene, naphthalene and phenanthreno are quite unreactive but anthracene responds readily.
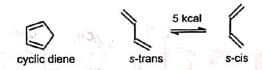
 The reaction occurs with the cisoid form (s-cis conformation) of the diene. Cyclic dienes are naturally in cisoid form and so react more rapidly than the acyclic dienes which normally exist in the more stable transoid forms (s-trans conformation).
The reaction occurs with the cisoid form (s-cis conformation) of the diene. Cyclic dienes are naturally in cisoid form and so react more rapidly than the acyclic dienes which normally exist in the more stable transoid forms (s-trans conformation).
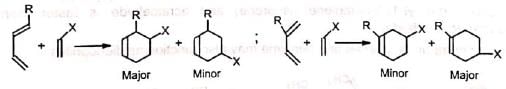
 When both the diene and dienophile are unsymmetrical, two products are possible. However, the 1, 2- and 1, 4-products predominate.
When both the diene and dienophile are unsymmetrical, two products are possible. However, the 1, 2- and 1, 4-products predominate.
Mechanism:
There is little evidence in favour of stepwise polar or free radical mechanism. The other possibility is a concerted mechanism. Both the mechanisms are discussed. However, see note below.
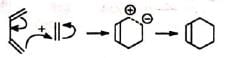
(i) Stepwise polar and free radical mechanism The mechanism envisages that the bonds between the reactants are formed consecutively, i.e., one bond is formed followed by the other. The first bond gives a diion (a) or a diradical (b) which then forms the second bond.
(a) Polar reaction
 (b) Free radical reaction
(b) Free radical reaction
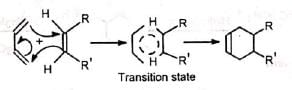
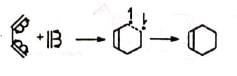 The mechanism assumes that the second bond is formed more rapidly than the rotation about the C-C bond, as otherwise a mixture of cis and trans products (nonstereospecific) would be obtained with substituted reactants, which is contrary to observation. The reaction is strictly stereospecific. The orientation of the groups (i.e., cis or trans) in the reactant remain unaltered in the product. Hence the mechanism is not favourable. (ii) Concerted mechanism The mechanism visualises a process in which there is simultaneous (not consecutive) breaking and making of bonds through a six-centred transition state with no intermediate. It is a 4π + 2π cycloaddition and stereospecifically cis with respect to both reactants.
The mechanism assumes that the second bond is formed more rapidly than the rotation about the C-C bond, as otherwise a mixture of cis and trans products (nonstereospecific) would be obtained with substituted reactants, which is contrary to observation. The reaction is strictly stereospecific. The orientation of the groups (i.e., cis or trans) in the reactant remain unaltered in the product. Hence the mechanism is not favourable. (ii) Concerted mechanism The mechanism visualises a process in which there is simultaneous (not consecutive) breaking and making of bonds through a six-centred transition state with no intermediate. It is a 4π + 2π cycloaddition and stereospecifically cis with respect to both reactants.
 The addition of the dienophile is always cis so that the groups cis in the olefin remain cis in the adduct. The addition is therefore stereospecific.
The addition of the dienophile is always cis so that the groups cis in the olefin remain cis in the adduct. The addition is therefore stereospecific.
There are two possible modes of addition, (a) exo and (b) endo. When the greater part of the dienophile is under the diene ring in the adduct, it is called endo and in the reverse case, it is called exo. The endo adduct is more stable.
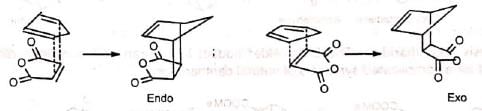 This is because the cyclic transition state has further stabilization through secondary 7i-orbital overlaps for greater accumulation of double bonds in the endo adduct.
This is because the cyclic transition state has further stabilization through secondary 7i-orbital overlaps for greater accumulation of double bonds in the endo adduct.
Applications:
The reaction has proved of great value. Due to its high stereospecific nature, the reaction has been used to synthesize many natural products which otherwise would be difficult to prepare. The reaction can be used as a diagnostic test for conjugation in a system. Since the reaction is stereospecific, the configurations of the reactants can be determined by studying the adduct. The reaction has also been used to trap benzyne intermediate. Some reactions are given to illustrate its use.
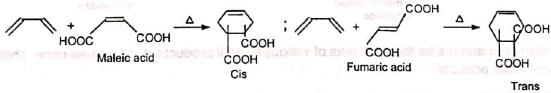
1. Determination of configuration Maleic and fumaric acids can be identified from the products.
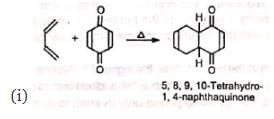
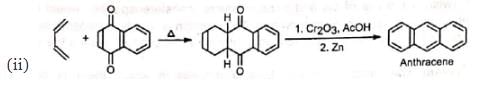
 2. Synthesis of quinones and polynuclear hydrocarbons
2. Synthesis of quinones and polynuclear hydrocarbons
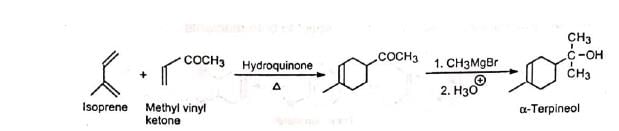
3. Synthesis of α-terpineol
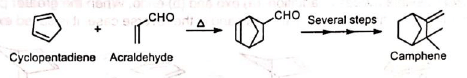
 4. In the synthesis of camphene The intermediate required in the synthesis of camphene is prepared by Diels-Alder reaction.
4. In the synthesis of camphene The intermediate required in the synthesis of camphene is prepared by Diels-Alder reaction.
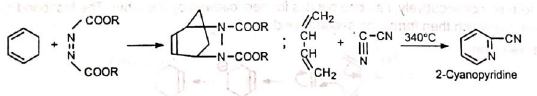
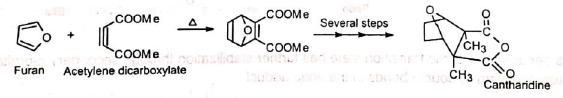
 5. In the synthesis of cantharidine The Diels-Alder adduct from furan and acetylene dicarboxylate is the starting product for a complicated synthesis of natural cantharidine.
5. In the synthesis of cantharidine The Diels-Alder adduct from furan and acetylene dicarboxylate is the starting product for a complicated synthesis of natural cantharidine.
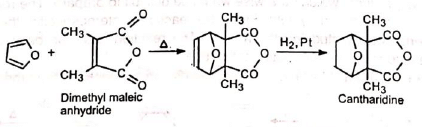
 The Diels-Alder adduct obtained from furan and dimethylmaleic anhydride on hydrogenation gives cantharidine which is not the same as the natural product.
The Diels-Alder adduct obtained from furan and dimethylmaleic anhydride on hydrogenation gives cantharidine which is not the same as the natural product.
 The starting materials for the synthesis of various natural products, such as reserpine, cholesterol, etc., are the Diels-Alder adducts.
The starting materials for the synthesis of various natural products, such as reserpine, cholesterol, etc., are the Diels-Alder adducts.
Note: The concerted cycloaddition is best explained from the principle of Conservation of Orbital Symmetry put forward by Woodward and Hoffmann. The principle states that cycloaddition is allowed when the symmetry properties of the highest occupied molecular orbital (HOMO) of one reactant correlates with the lowest unoccupied molecular orbital (LUMO) of the other and vice versa.
It is seen that in the reactants in the ground state, the signs of 1 , 4-lobes of lumo for 1, 3-butadiene correlates with those of homo of ethylene, and hence the addition occurs smoothly on heating.
(Orbitals of the same signs, i.e., of same phase only overlap to form bonds.)
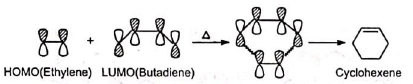 Similar correlation is observed between butadiene homo and ethylene lumo.
Similar correlation is observed between butadiene homo and ethylene lumo.
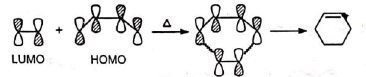 The stereospecificity (i.e., cis-addition) is thus explained.
The stereospecificity (i.e., cis-addition) is thus explained.



