Cellular Biochemistry


Cell Cycle and Checkpoints
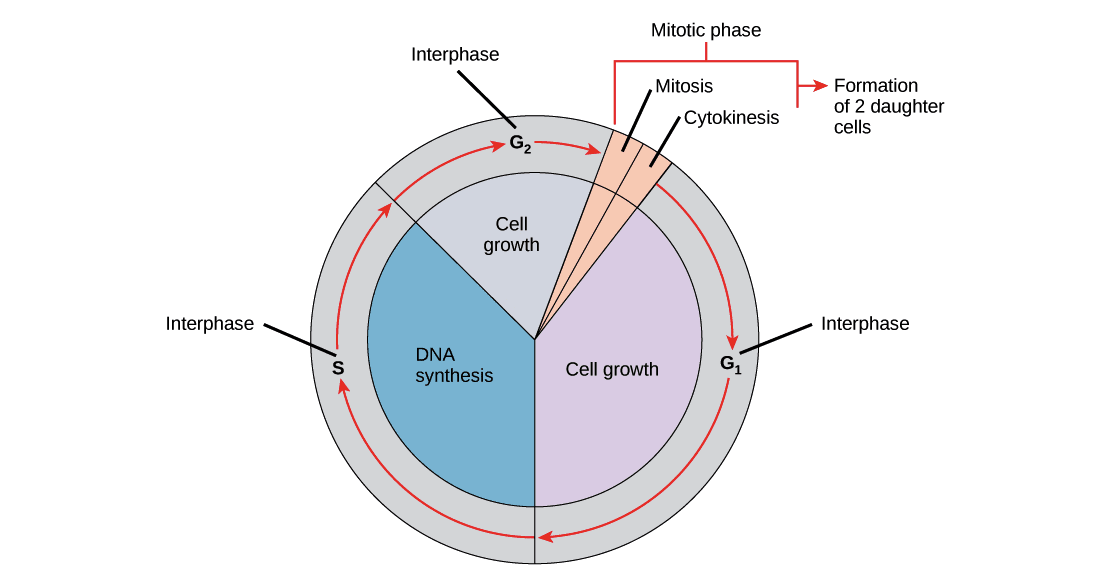
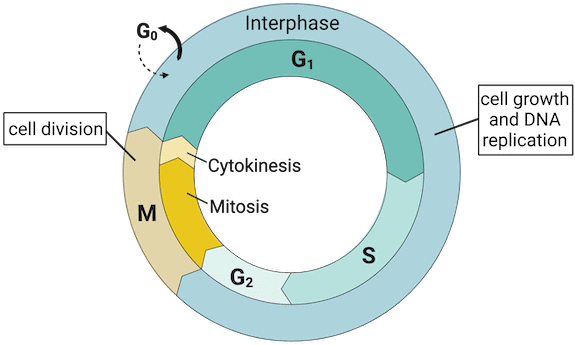
The cell cycle is the ordered series of events by which a cell duplicates its contents and divides. The main phases are:
- G0: A quiescent state in which cells have left the cell cycle and are not actively dividing.
- G1 (first gap): Cell grows, synthesises proteins and organelles; duration is variable and is the phase during which the cell decides whether to divide.
- S (synthesis): DNA replication occurs; each chromosome is duplicated to form sister chromatids.
- G2 (second gap): Further growth and preparation for mitosis; DNA repair mechanisms check replication completeness.
- M (mitosis): Nuclear division, followed by cytokinesis (division of the cytoplasm). Mitosis is subdivided into prophase, prometaphase, metaphase, anaphase and telophase.
Checkpoints at the G1/S and G2/M boundaries and during metaphase ensure DNA integrity, completion of DNA replication and correct chromosome attachment to the spindle. These checkpoints prevent progression if conditions are unfavourable.
Regulators of the Cell Cycle
Progression through the cell cycle is tightly controlled by interacting molecules and pathways.
Cyclins and Cyclin-Dependent Kinases (CDKs)
- CDKs are protein kinases that are constitutively present but inactive unless bound to a specific cyclin.
- Cyclins are regulatory proteins whose levels rise and fall according to the cell cycle phase.
- When a cyclin binds a CDK, the resulting cyclin-CDK complex phosphorylates target proteins to drive phase-specific events (for example, phosphorylation of the retinoblastoma protein Rb to permit S-phase entry).
- Activation and inactivation of cyclin-CDK complexes at the correct times are essential for orderly cell-cycle progression.
Tumour Suppressors and CDK Inhibitors
- p53 is a transcription factor that responds to DNA damage.
- Activated p53 induces expression of the CDK inhibitor p21, which inhibits cyclin-CDK activity and causes cell-cycle arrest (commonly at the G1/S checkpoint) to allow DNA repair or to trigger apoptosis if damage is irreparable.
- Loss of p53 function leads to failure of this checkpoint and increased risk of malignancy (for example, germline p53 mutations cause Li-Fraumeni syndrome).
- Rb (retinoblastoma protein) restrains the G1→S transition by binding and inhibiting E2F transcription factors.
- Phosphorylation of Rb by cyclin-CDK complexes releases E2F, allowing transcription of genes required for S phase.
- Hypophosphorylated (active) Rb thus prevents S-phase entry.
Growth Factor Signalling
Mitogenic growth factors (for example, insulin, platelet-derived growth factor PDGF, erythropoietin EPO, epidermal growth factor EGF) bind receptor tyrosine kinases and activate intracellular signalling cascades that promote cyclin expression and G1→S transition.
Cell-Cycle Arrest and Apoptosis Link
Persistent DNA damage can lead to p53-mediated upregulation of BAX and related pro-apoptotic proteins, triggering the intrinsic (mitochondrial) apoptotic pathway with caspase activation. Anti-apoptotic proteins such as BCL-2 and BCL-XL oppose this process.
Cell Types and Proliferative Capacity
Different cell types vary in their ability to divide:
- Permanent (terminally differentiated) cells: Remain in G0 and do not normally regenerate; they are replenished only from stem cells. Examples: neurons, skeletal and cardiac muscle cells, erythrocytes.
- Stable (quiescent) cells: Normally in G0 but can re-enter G1 in response to stimuli. Examples: hepatocytes, lymphocytes, proximal tubule cells, periosteal cells.
- Labile cells: Continuously proliferating and rarely enter G0; they have a short G1 and are most sensitive to cytotoxic chemotherapy. Examples: bone marrow progenitors, gut epithelium, skin, hair follicles, germ cells.
Rough Endoplasmic Reticulum (RER)
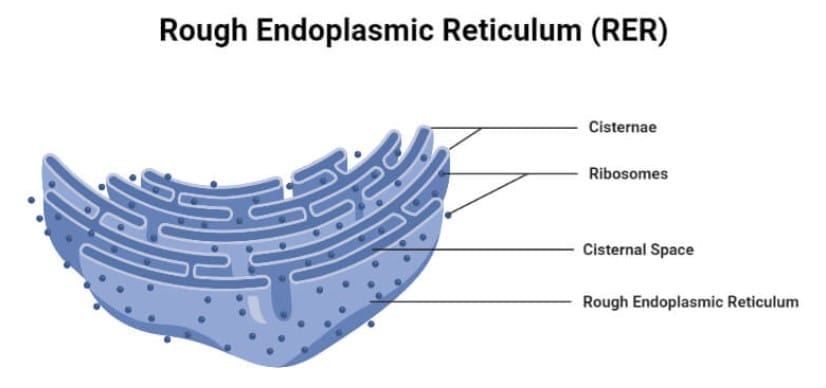
- RER is studded with ribosomes and is the site of synthesis of secreted and membrane-bound proteins.
- It is where N-linked glycosylation (addition of oligosaccharides to asparagine residues) begins.
- Cells specialised for protein secretion (for example, antibody-secreting plasma cells and mucus-secreting goblet cells) have extensive RER. In neurons, RER appears as Nissl bodies and is involved in peptide neurotransmitter synthesis.
- Free ribosomes in the cytosol synthesise proteins that remain in the cytosol, are targeted to peroxisomes or mitochondria, or are imported into the nucleus.
Smooth Endoplasmic Reticulum (SER)
- SER lacks ribosomes and is involved in lipid and steroid synthesis, detoxification of drugs and poisons, and carbohydrate metabolism (for example, it contains glucose-6-phosphatase - the final step of gluconeogenesis and glycogenolysis).
- Hepatocytes and steroid-producing cells of the adrenal cortex and gonads are rich in SER.
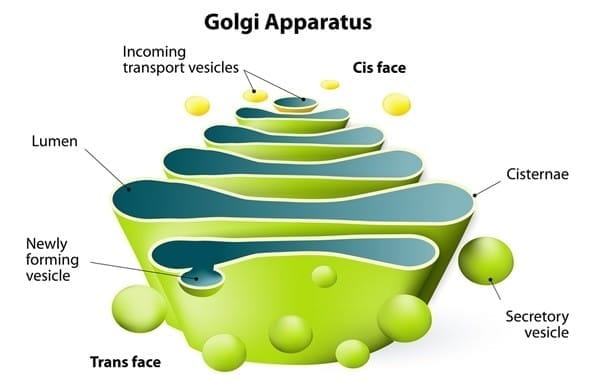
Protein Trafficking: Golgi, Endosomes and Sorting
The Golgi apparatus is the central sorting and modification station for proteins and lipids arriving from the ER. Post-translational modifications occurring in the Golgi include:
- Modification of N-linked oligosaccharides.
- Addition of O-linked oligosaccharides to serine and threonine residues.
- Addition of mannose-6-phosphate (M6P) tags that target hydrolases to lysosomes.
Endosomes sort material internalised by endocytosis or routed from the Golgi; they direct cargo to lysosomes for degradation or recycle it back to the plasma membrane or Golgi.
Vesicular Trafficking Machinery
- Signal recognition particle (SRP): A cytosolic ribonucleoprotein that recognises signal peptides on nascent polypeptides and directs the ribosome-polypeptide complex to the RER. Defects in SRP impair targeting to the ER and cause cytosolic accumulation of proteins.
- COPII: Mediates anterograde transport from the ER to the cis-Golgi. Mnemonic: "Two steps forward."
- COPI: Mediates retrograde transport from Golgi back to ER and from trans-Golgi toward cis-Golgi. Mnemonic: "One step back."
- Clathrin: Coats vesicles between the trans-Golgi network and endosomes/lysosomes, and mediates receptor-mediated endocytosis at the plasma membrane (for example, LDL receptor internalisation).
Diseases of Golgi/Lysosomal Targeting: I-Cell Disease
- I-cell disease (inclusion-cell disease, mucolipidosis type II) is an autosomal recessive disorder caused by deficiency of the enzyme N-acetylglucosaminyl-1-phosphotransferase, which catalyses the first step in formation of mannose-6-phosphate tags on lysosomal hydrolases. Without M6P tags, lysosomal enzymes are secreted extracellularly instead of being delivered to lysosomes, resulting in accumulation of undegraded substrates within lysosomes (inclusion bodies).
- Clinical features include coarse facial features, gingival hypertrophy, corneal clouding, restricted joint mobility, claw-hand deformities, kyphoscoliosis and elevated plasma levels of lysosomal enzymes. The disorder is severe and often fatal in childhood.
Peroxisomes
Peroxisomes are membrane-bound organelles that perform oxidative reactions. Key functions include:
- α-oxidation and β-oxidation of very-long-chain and branched-chain fatty acids (partially or completely peroxisomal processes).
- Catabolism of amino acids and ethanol.
- Synthesis of bile acids.
- Synthesis of plasmalogens, specialised phospholipids important for myelin and other membranes.
Peroxisomal disorders:
- Zellweger spectrum (peroxisome biogenesis disorders) - autosomal recessive mutations in PEX genes cause failure of peroxisome formation, leading to accumulation of very-long-chain fatty acids and other toxic metabolites. Clinical features include hypotonia, seizures, craniofacial dysmorphism, hepatomegaly and early death.
- Refsum disease - defective α-oxidation leading to accumulation of phytanic acid. Features include retinitis pigmentosa, anosmia, hearing loss, ataxia, peripheral neuropathy, ichthyosis and cardiac conduction defects. Treatment: dietary restriction of phytanic acid and plasmapheresis in severe cases.
- Adrenoleukodystrophy (X-linked) - mutation in the ABCD1 gene impairs peroxisomal β-oxidation of very-long-chain fatty acids, causing accumulation in adrenal glands, white matter and testes. Progressive demyelination and adrenal insufficiency are typical; presentation and severity vary.
Proteasome
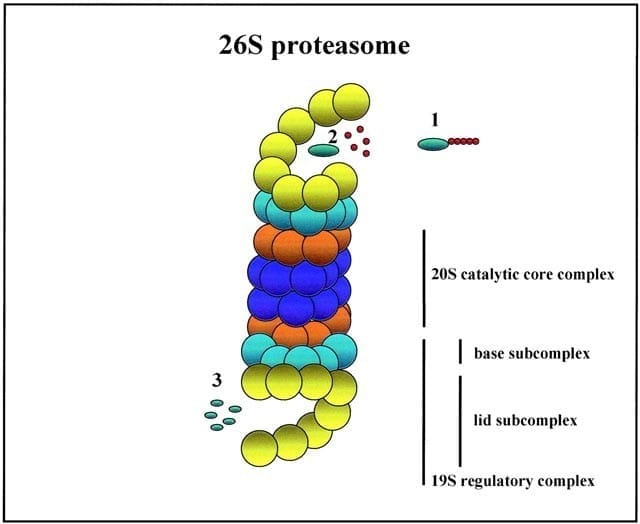
- The ubiquitin-proteasome system tags proteins with polyubiquitin chains for degradation by the proteasome, a barrel-shaped complex.
- This system controls protein quality and regulates cellular processes such as cell-cycle progression and the immune response (for example, production of peptides for MHC class I presentation).
- Proteasome dysfunction has been implicated in neurodegenerative diseases and other pathologies.
Cytoskeleton
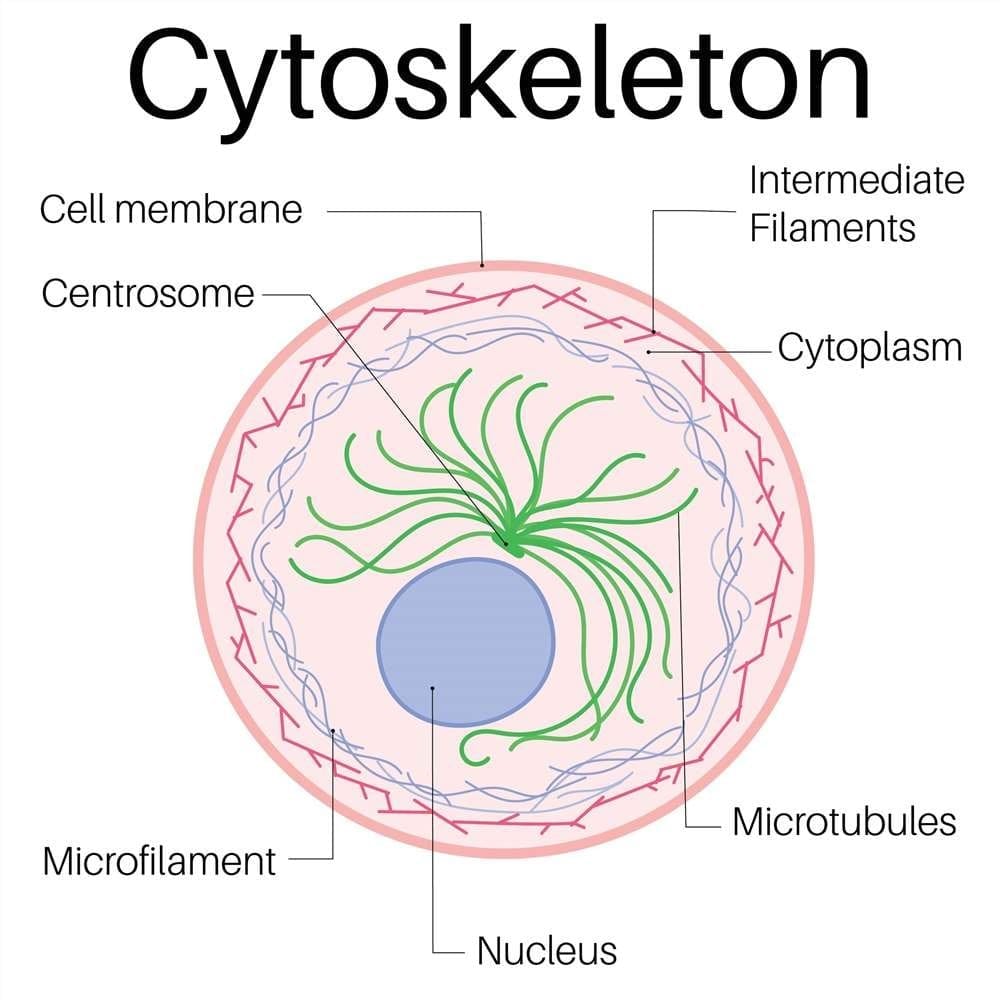
The cytoskeleton is composed of three main filament systems with distinct functions: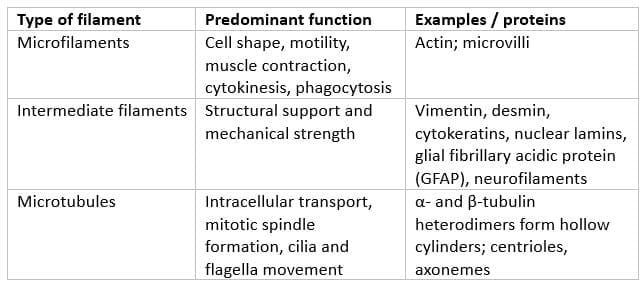
Microtubule Structure and Polarity
- Microtubules are cylindrical polymers of α- and β-tubulin heterodimers arranged into protofilaments.
- Each tubulin dimer binds two molecules of GTP (one on α-tubulin is non-exchangeable; one on β-tubulin is hydrolysed during polymerisation).
- Microtubules are polar structures with a fast-growing plus (+) end usually oriented toward the cell periphery, and a slower-growing minus (-) end oriented toward the microtubule organising centre near the nucleus.
Molecular Motors
- Dynein: A minus-end-directed motor protein that mediates retrograde transport (from cell periphery toward the nucleus).
- Kinesin: Generally a plus-end-directed motor that mediates anterograde transport (from the nucleus toward the periphery).
Many viruses exploit these motors for intracellular transport: for example, rabies virus, poliovirus and herpes simplex virus (HSV) use dynein for retrograde transport to the neuronal cell body. HSV particles are transported anterogradely by kinesin for reactivation and spread.
Drugs that Affect Microtubules
- Colchicine: Binds tubulin and inhibits microtubule polymerisation; used in gout.
- Vincristine, vinblastine (vinca alkaloids): Inhibit microtubule polymerisation; used as anticancer agents.
- Taxanes (for example paclitaxel): Stabilise microtubules and prevent disassembly; used in cancer chemotherapy.
- Mebendazole: An anthelmintic that inhibits tubulin polymerisation in parasites.
- Griseofulvin: An antifungal that disrupts microtubule function and mitosis.
Cilia and Motility
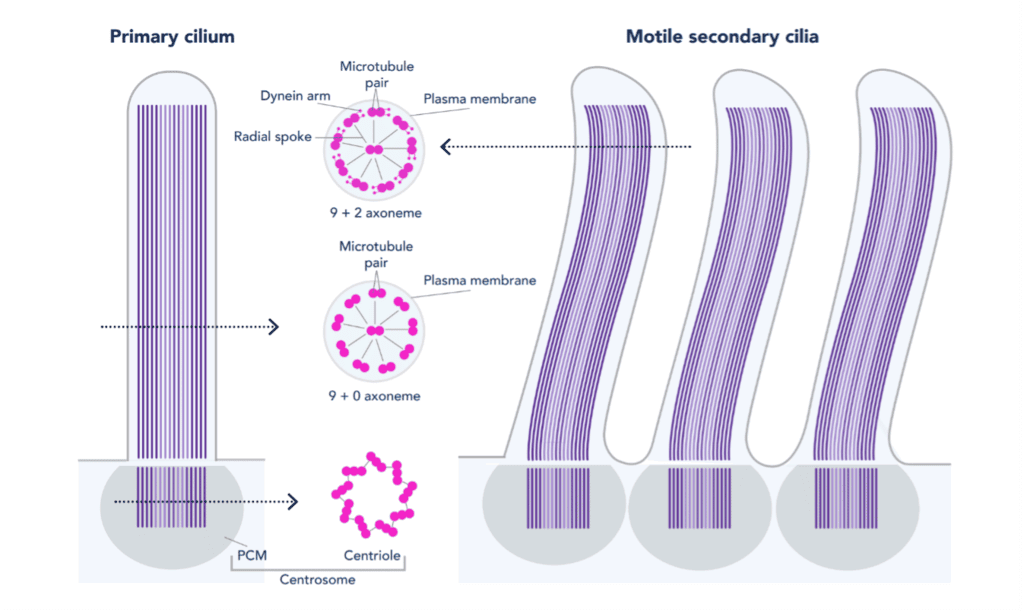
Structure of Motile Cilia
- Motile cilia have an axoneme with a characteristic "9+2" arrangement: nine peripheral microtubule doublets surrounding two central singlet microtubules. The basal body that anchors the cilium contains nine microtubule triplets and lacks the central microtubules.
- Axonemal dynein arms are ATPase motor proteins that generate sliding between adjacent doublets, producing bending and motility. Nexin links between doublets restrict sliding and convert it into bending.
Primary (Non-Motile) Cilia
- Primary cilia have a "9+0" microtubule arrangement and act as sensory organelles involved in signal transduction and control of cell growth.
- Defects in primary cilia can contribute to developmental disorders such as polycystic kidney disease and retinal degeneration.
Primary Ciliary Dyskinesia and Kartagener Syndrome
- Primary ciliary dyskinesia (PCD) is usually an autosomal recessive disorder caused by defects in dynein arms or other axonemal components, producing immotile or dyskinetic cilia. Consequences include chronic sinusitis, recurrent otitis media, bronchiectasis due to impaired mucociliary clearance, and infertility (immotile sperm; dysfunctional fallopian tube cilia, increasing risk of ectopic pregnancy).
- Kartagener syndrome is a subset of PCD with the triad of situs inversus, chronic sinusitis and bronchiectasis, resulting from abnormal left-right body axis determination due to defective ciliary motility during embryogenesis.
- Screening can include measurement of nasal nitric oxide, which is often low in PCD.
Membrane Transport: Na+/K+ ATPase
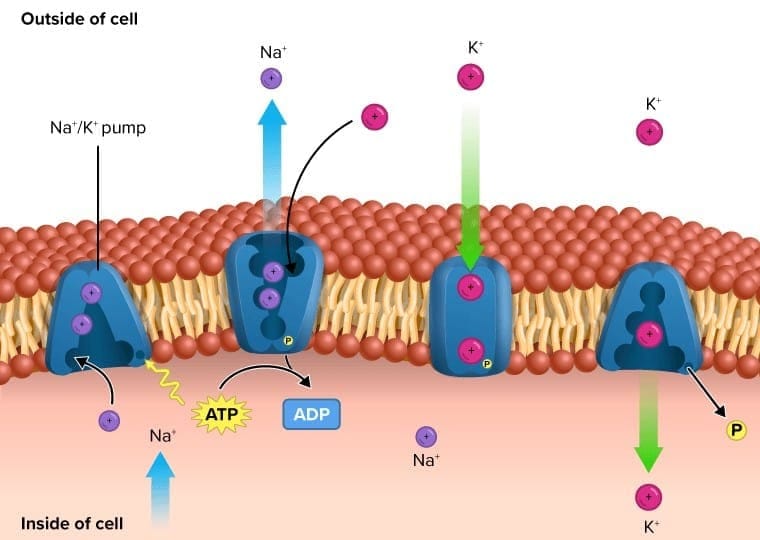
- The Na+/K+-ATPase is an integral plasma membrane enzyme that uses ATP to transport ions against their concentration gradients. For each ATP hydrolysed, the pump exports three Na+ ions and imports two K+ ions, contributing to the resting membrane potential and maintaining cellular ionic homeostasis.
- Cardiac glycosides such as digoxin inhibit the Na+/K+-ATPase. Inhibition increases intracellular Na+, which secondarily reduces the activity of the Na+/Ca2+ exchanger, causing intracellular Ca2+ accumulation and increased cardiac contractility.
Collagen: Types, Synthesis and Disorders
- Collagen is the most abundant protein in the human body, forming a major component of the extracellular matrix and conferring tensile strength to tissues.
- Collagens are extensively post-translationally modified. The most commonly discussed human types are types I-IV.
Common Collagen Types and Locations (Mnemonic: SCAB)
- Type I: Bone, skin, tendon, dentin, fascia, cornea and late wound repair (makes up ~90% of body collagen).
- Type II: Cartilage (including hyaline cartilage), vitreous body, nucleus pulposus.
- Type III: Reticular fibres in skin, blood vessels, uterus, fetal tissue and early wound repair (reticulin).
- Type IV: Basement membrane and basal lamina (for example renal glomerular basement membrane, lens).
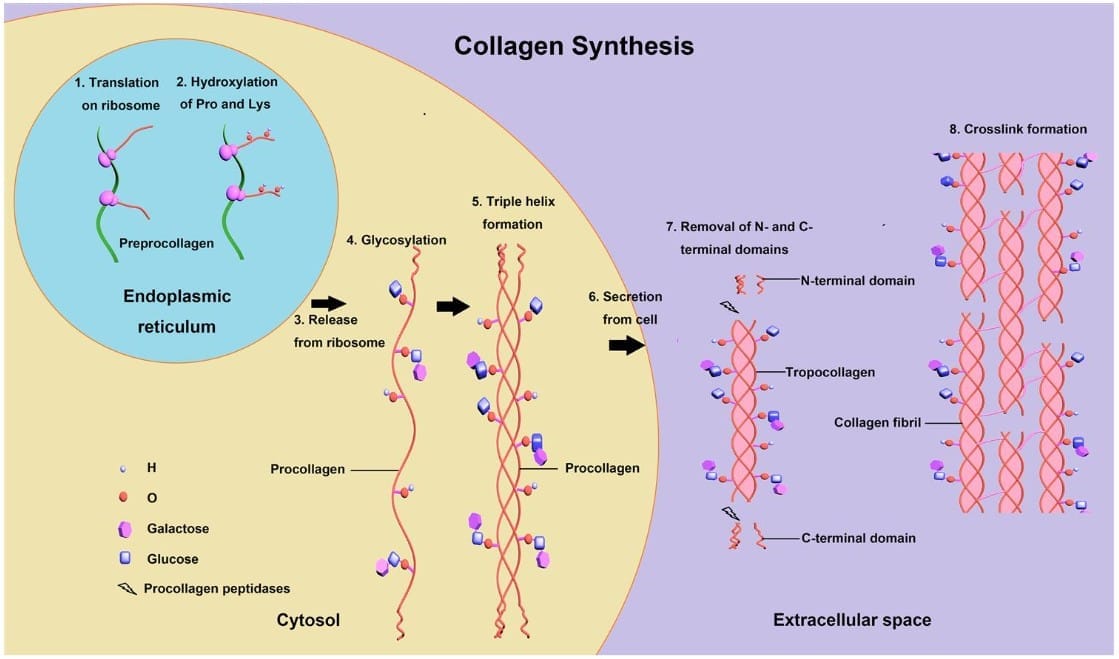
Collagen Synthesis and Post-Translational Modifications
Key steps in collagen biosynthesis:
- Translation: Synthesis of preprocollagen α chains on ribosomes; typical repeating sequence is Gly-X-Y where X and Y are often proline or lysine (approximately one-third of residues are glycine).
- Hydroxylation: Specific proline and lysine residues are hydroxylated by prolyl and lysyl hydroxylases; this requires vitamin C (ascorbic acid). Deficiency leads to scurvy with impaired collagen stability.
- Glycosylation: Glycosylation of hydroxylysine residues and formation of procollagen via hydrogen and disulfide bonds; triple helix (procollagen) formation depends on these modifications. Defects in triple-helix formation contribute to osteogenesis imperfecta.
- Secretion: Procollagen is secreted by exocytosis into the extracellular space.
- Proteolytic processing: Extracellular cleavage of N- and C-terminal propeptides produces insoluble tropocollagen.
- Cross-linking: Lysyl oxidase (a copper-dependent enzyme) oxidatively deaminates lysine and hydroxylysine residues to allow covalent cross-links between tropocollagen molecules, forming strong collagen fibres. Defects in lysyl oxidase lead to weak cross-linking (for example, in Menkes disease).
Osteogenesis Imperfecta
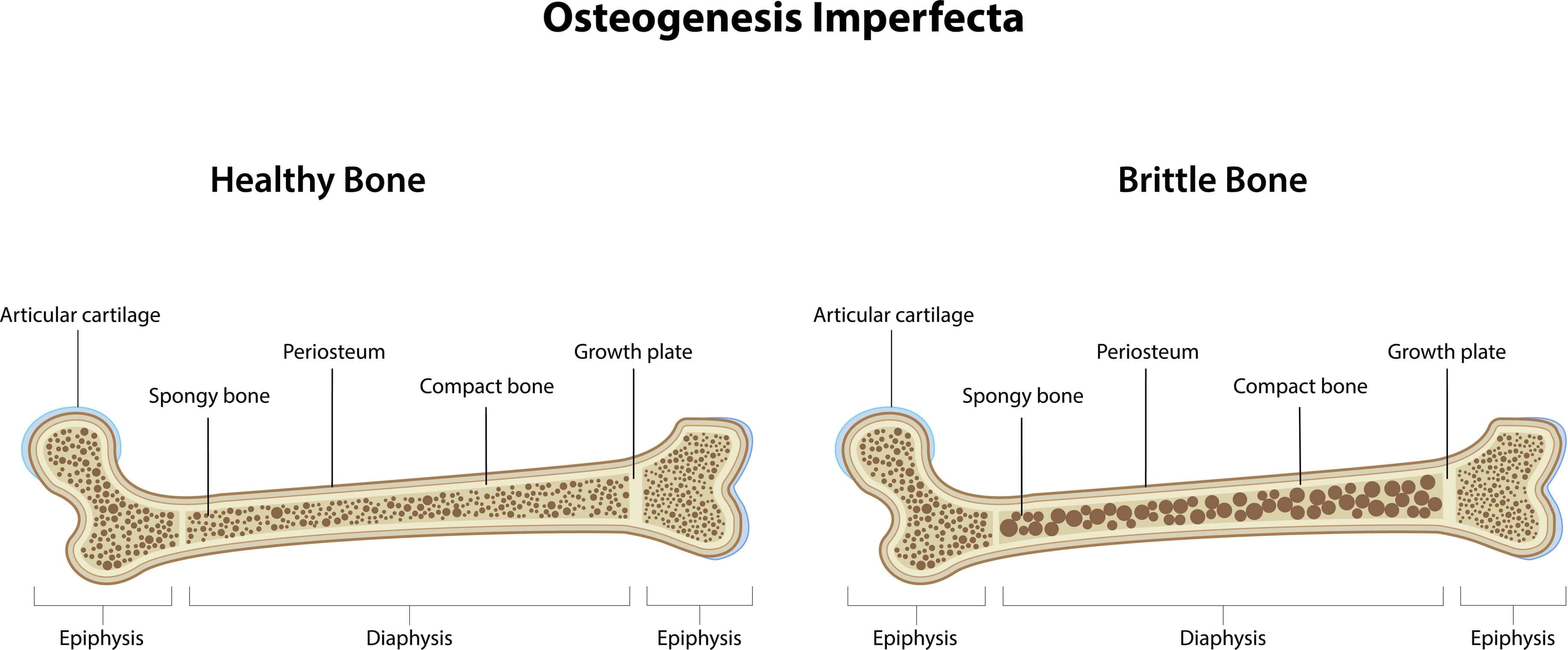
- Osteogenesis imperfecta (brittle bone disease) is most commonly caused by autosomal dominant mutations in COL1A1 and COL1A2, genes encoding type I collagen.
- Many mutations reduce the amount or impair the structure of type I collagen.
- Clinical features include multiple fractures with minimal trauma, bone deformities, blue sclerae (thin sclera revealing underlying choroidal veins), dentinogenesis imperfecta (opalescent, brittle teeth), and hearing loss due to abnormal ossicles.
- Treatment is supportive and may include bisphosphonates to reduce fracture risk.
Mnemonic: Patients with osteogenesis imperfecta can't BITE - Bones (fractures), I (eye: blue sclerae), Teeth (dentinogenesis imperfecta), Ear (hearing loss).
Ehlers-Danlos Syndromes
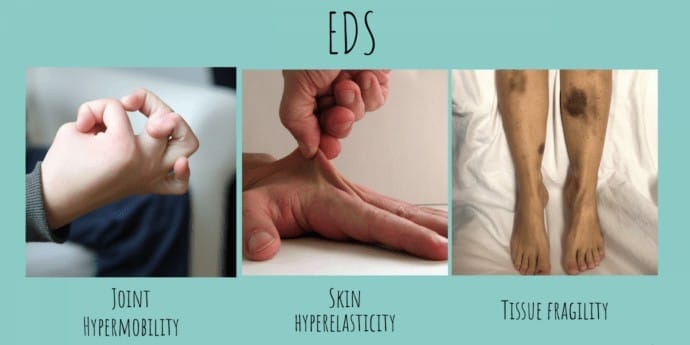
Ehlers-Danlos syndromes (EDS) are a group of disorders of connective tissue characterised by hyperextensible skin, hypermobile joints and tissue fragility with easy bruising. There are multiple genetic subtypes with variable inheritance.
- Classical type: Often due to mutations in type V collagen (COL5A1, COL5A2); features: skin hyperextensibility and joint hypermobility.
- Vascular type: Due to defects in type III collagen (COL3A1); features include fragile vessels and hollow organs with risk of rupture, thin translucent skin and easy bruising.
- Hypermobility type: Primarily joint instability and pain; most common type.
Some forms arise from defects in enzymes that process procollagen (for example, procollagen peptidase deficiency).
Menkes Disease
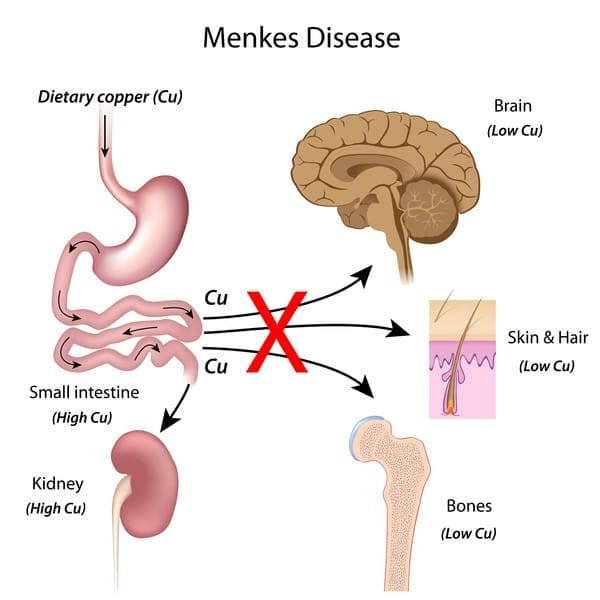
- Menkes disease is an X-linked recessive disorder caused by mutations in the ATP7A copper-transporting ATPase leading to impaired intestinal copper absorption and defective copper distribution.
- Copper-dependent enzymes such as lysyl oxidase have reduced activity, impairing collagen cross-linking. Clinical features include growth and developmental delay, hypotonia, brittle, kinky (pili torti) hair, and a tendency to vascular abnormalities.
- This contrasts with Wilson disease (mutations in ATP7B) where copper accumulates rather than being deficient.
Summary
This chapter covered the essentials of cellular biochemistry relevant to cell division, organelle function, intracellular trafficking, cytoskeletal structure and dynamics, membrane transport and extracellular matrix biology. Understanding the molecular players (cyclins and CDKs, tumour suppressors, trafficking machinery, peroxisomal and proteasomal systems, microtubule motors, ion pumps and collagen biosynthetic enzymes) explains how normal cellular function is achieved and how defects produce classical clinical syndromes.
FAQs on Cellular Biochemistry
| 1. What are the key phases of the cell cycle and their significance? |  |
| 2. What role do checkpoints play in the cell cycle? | |
| 3. How do collagen types differ in structure and function? | |
| 4. What is the function of the proteasome in cellular processes? | |
| 5. How does the Na⁺/K⁺ ATPase contribute to cellular homeostasis? | |


