Bonding in Coordination Compounds: VBT and CFT | Chemistry Class 12 - NEET PDF Download

What is Valence Bond Theory(VBT)?
According to the Valence Bond Theory, electrons in a molecule occupy atomic orbitals rather than molecular orbitals. The atomic orbitals overlap on the bond formation and the larger the overlap the stronger the bond.
The metal bonding is essentially covalent in origin and the metallic structure involves the resonance of electron-pair bonds between each atom and its neighbours.
History of Valence Bond Theory
The Lewis approach to chemical bonding failed to shed light on the formation of chemical bonds. Also, valence shell electron pair repulsion theory (or VSEPR theory) had limited applications (and also failed in predicting the geometry corresponding to complex molecules).
In order to address these issues, the valence bond theory was put forth by the German physicists Walter Heinrich Heitler and Fritz Wolfgang London. The Schrodinger wave equation was also used to explain the formation of a covalent bond between two hydrogen atoms. The chemical bonding of two hydrogen atoms as per the valence bond theory is illustrated below.
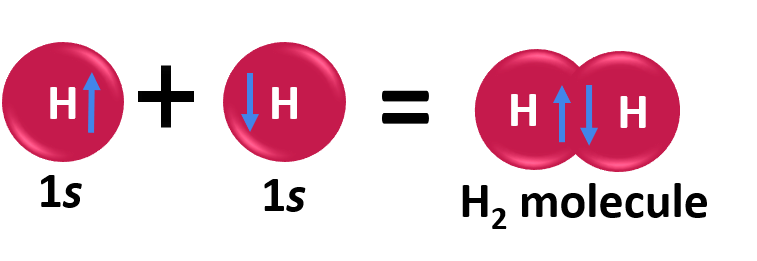
This theory focuses on the concepts of electronic configuration, atomic orbitals (and their overlapping) and the hybridization of these atomic orbitals. Chemical bonds are formed from the overlapping of atomic orbitals wherein the electrons are localized in the corresponding bond region.
The valence bond theory also goes on to explain the electronic structure of the molecules formed by this overlapping of atomic orbitals. It also emphasizes that the nucleus of one atom in a molecule is attracted to the electrons of the other atoms.
Postulates of Valence Bond Theory
It was developed by Pauling. The salient features of the theory are summarised below :
- Under the influence of strong-field ligands, the electrons of the central metal ion can be forced to pair up against Hund's rule of maximum multiplicity.
- Under the influence of weak field ligands, the electronic configuration of central metal atom and ion remains the same.
- If the complex contains unpaired electrons, it is paramagnetic in nature, whereas if it does not contain unpaired electrons, then it is diamagnetic in nature and magnetic moment is calculated by spin only formula.
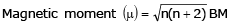 where n is the number of unpaired electrons in the metal ion.
where n is the number of unpaired electrons in the metal ion.
Relation between unpaired electrons and magnetic momentMagnetic moment (Bohr magnetons)
0
1.73
2.83
3.87
4.90
5.92
Number of unpaired electrons
0
1
2
3
4
5
Thus, the knowledge of the magnetic moment can be of great help in ascertaining the type of complexity. - When ligands are arranged in increasing order of their splitting power then an experimentally determine series is obtained named as spectrochemical series.


- The central metal ion has a number of empty orbitals for accommodating electrons donated by the ligands.
The number of empty orbitals is equal to the coordination number of the metal ion for a particular complex. - The atomic orbital (s, p or d) of the metal ion hybridise to form hybrid orbitals with definite directional properties. These hybrid orbitals now accept e- pairs from ligands to form coordination bonds.
- The d-orbitals involved in the hybridisation may be either inner (n - 1) d orbitals or outer n d-orbitals. The complexes formed in these two ways are referred to as inner orbital complexes and outer orbital complexes, respectively.
Number of Orbitals and Types of Hybridization
According to VBT theory the metal atom or ion under the influence of ligands can use its (n-1)d, ns, np, or ns, np, nd orbitals for hybridization to yield a set of equivalent orbitals of definite geometry such as octahedral, tetrahedral, square planar and so on. These hybrid orbitals are allowed to overlap with ligand orbitals that can donate electron pairs for bonding.
| Coordination Number | Type of Hybridisation | Distribution of Hybrid Orbitals in Space |
| 4 | sp3 | Tetrahedral |
| 4 | dsp2 | Square planar |
| 5 | sp3d | Trigonal bipyramidal |
| 6 | sp3d2 | Octahedral |
| 6 | d2sp3 | Octahedral |
Limitations of Valence Bond Theory
- Correct magnetic moment of complex compounds can not be theoretically measured by Valence bond theory.
- The theory does not offer any explanation about the spectra of complex (i.e., why most of the complexes are coloured).
- Theory does not offer any explanation for the existence of inner -orbital and outer -orbital complexes.
- In the formation of [Cu(NH3)4]2+, one electron is shifted from 3d to 4p orbital. The theory is silent about the energy availability for shifting such an electron.
Such an electron can be easily lost then why does not [Cu(NH3)4]2+ complex show reducing properties.
Applications of Valence Bond Theory
- The maximum overlap condition which is described by the valence bond theory can explain the formation of covalent bonds in several molecules.
- This is one of its most important applications. For example, the difference in the length and strength of the chemical bonds in H2 and F2 molecules can be explained by the difference in the overlapping orbitals in these molecules.
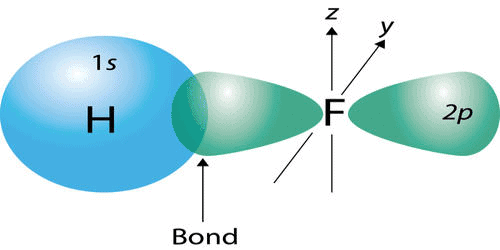
- The covalent bond in an HF molecule is formed from the overlap of the 1s orbital of the hydrogen atom and a 2p orbital belonging to the fluorine atom, which is explained by the valence bond theory.
Magnetic Properties of Complexes
The complex in which central transition metal ion has unpaired electrons is Paramagnetic. The complex in which central transition metal ion has no unpaired electrons is Diamagnetic.
The magnetic moment of a complex is calculated by the spin only formula
- M = √[n(n+2)] BM
- BM = Bohr Magneton
The magnetic moment of complex compounds depends upon:
- Type of hybridization.
- The oxidation state of central transition metal ion.
- The number of unpaired electrons.
What is Crystal Field Theory?
Crystal field theory describes that the net change in crystal energy resulting from the orientation of d orbitals of a transition metal cation inside a coordinating group of anions also called ligands.

- Crystal field theory is now much more widely accepted than the valence bond theory. It is assumed that the attraction between the central metal and ligands in a complex is purely electrostatic.
- The transition metal which forms the central atom cation in the complex is regarded as a positive ion of charge equal to the oxidation state.
- It is surrounded by negative ligands or neutral molecules which have a lone pair of electrons If the ligand is a neutral molecule such as NH3, the negative end of the dipole in the molecule is directed toward the metal cation.
- The electrons on the central metal are under repulsive forces from those on the ligands. Thus, the electrons that occupy the d-orbital remain away from the direction of the approach of ligands.
Overview of Crystal Field Theory
In the crystal field theory, the following assumptions are made:
(i) Ligands are treated as point charges.
(ii) There is no interaction between metal orbitals and ligands orbitals.
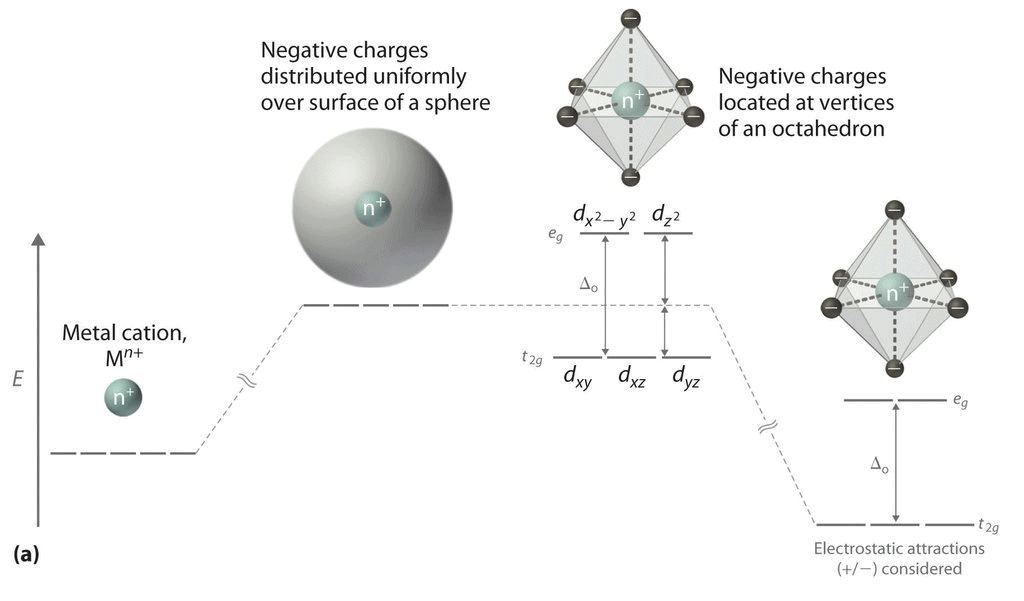
(iii) All the d orbitals on the metal have the same energy (that, is degenerate) in the free atom. However, when a complex, is formed, the ligands destroy the degeneracy of these orbitals, that is, the orbitals now have different energies. In an isolated gaseous metal ion, all five d orbitals have the same energy and are termed degenerate. If a spherically symmetrical field of ligands surrounds the metal ion, the d orbitals remain degenerate. (iv) However, the energy of the orbitals is raised because of repulsion between the field of ligands and electrons on the metal. In most transition metal complexes, either six or four ligands surround the metal, giving octahedral or tetrahedral structures. In both these cases, the field produced by the ligands is not spherically symmetrical. Thus, the d orbitals are not all affected equally by the ligand field.
High Spin and Low Spin
The complexion with the greater number of unpaired electrons is known as the high spin complex, the low spin complex contains the lesser number of unpaired electrons. High spin complexes are expected with weak field ligands whereas the crystal field splitting energy is small Δ. The opposite applies to the low spin complexes in which strong field ligands cause maximum pairing of electrons in the set of three t2 atomic orbitals due to large Δo.
- High spin – Maximum number of unpaired electrons.
- Low spin – Minimum number of unpaired electrons.
Example:
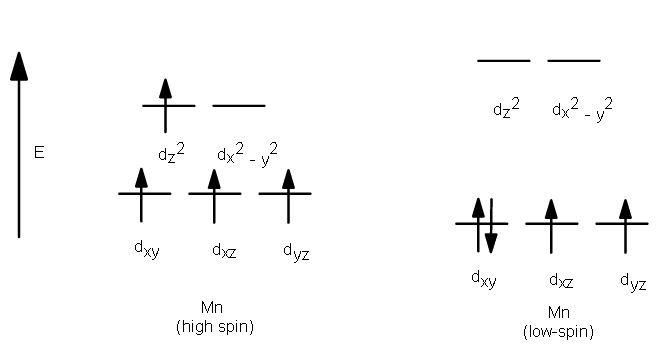
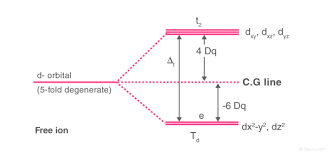
Crystal Field Splitting in Octahedral Complex
In the octahedral complex, the metal is at the centre of the octahedron and the ligands are at the six corners.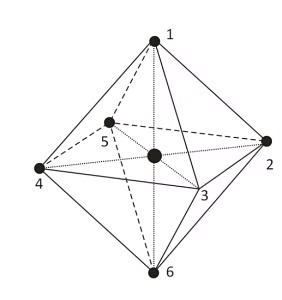
The direction x, y and z point to three adjacent corners of the octahedron.
The lobes of the eg orbitals (dx2-y2 and dz2) point along the axes x,y and z. The lobes of the t2g orbitals (dxy, dxz and dyz) point in between the axes. If follows that the approach of six ligands along the x,y,z, -x, -y, and -z directions will increase the energy of the dx2 - y2 and dz2 orbitals (which point along the axes) then it increases the energy of the dxy, dxz and dyz orbitals (which points between the axes). Thus, under the influence of an octahedral ligand field, the d orbitals split into two groups of different energies.
Rather than referring to the energy level of an isolated metal atom. The difference in energy between the two d levels is given by the symbols Δ0 or 10 Δq.
It follows that the eg orbitals are 0.6 Δ0 above the average level, and the t2g orbitals -0.4 Δ0 below the average level.
Crystal Field Splitting in Tetrahedral Complex
A regular tetrahedron is related to a cube. One atom is at the centre of the cube, and four of the eight corners of the cube are occupied by ligands as shown in Fig. The direction x,y and z point to the centres of the faces of the cube. The eg orbitals point along x,y and z (that is, to centres of the faces.)
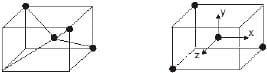 Relation of the tetrahedron to a cube
Relation of the tetrahedron to a cube
The approach of the ligands raised the energy of both sets of orbitals. The energy of the t2g orbital raised most because they are closest to the ligands. This crystals field splitting is opposite to that in octahedral complexes. The t2g orbitals are 0.4 Δt above the average energy of the two groups (the barycenter) and the eg orbitals are 0.6 Δt below the average level.
Limitations Of Crystal Field Theory
The crystal field theory is highly useful and more significant as compared to the valence bond theory. Even after such useful properties, it has many limitations. The following points will clearly state the limitations of crystal field theory:
- The assumption that the interaction between metal-ligand is purely electrostatic cannot be said to be very realistic
- This theory takes only the d-orbitals of a central atom into account. The s and p orbits are not considered for the study
- The theory fails to explain the behaviour of certain metals which cause large splitting while others show small splitting. For example, the theory has no explanation as to why H2O is a stronger ligand as compared to OH–
- The theory rules out the possibility of having p bonding. This is a serious drawback because is found in many complexes.
- The theory gives no significance to the orbits of the ligands. Therefore, it cannot explain any properties related to ligand orbitals and their interaction with metal orbitals.
Crystal Field Stabilization Energy(CFSE)
In a chemical environment, the energy levels generally split as directed by the symmetry of the local field surrounding the metal ion. The energy difference between the eg and t2g levels is given as or 10Dq. It states that each electron that goes into the lower t2g level stabilizes the system by an amount of -4Dq and the electron that goes into eg level destabilizes the system by +6Dq. That is the t2g is lowered by 4Dq and the eg level is raised by +6Dq.Example: The net change in energy for d5 and d10 systems will be zero as shown below.
d5 :- 3(-4Dq) + 2(+6Dq) = -12Dq + 12Dq = 0
d10 :- 6(-4Dq) + 4(+6Dq) = -24Dq + 24Dq = 0
The decrease in energy caused by the splitting of the energy levels is called the “Ligand Field Stabilization Energy (LFSE)”.
Crystal Field Stabilization Energy Table
| Electronic Configuration | Octahedral Complex | Tetrahedral Complex | ||
| Weak Field (-Dq) | Strong Field (-Dq) | Weak Field (-Dq) | Strong Field (-Dq) | |
| d0 | 0 | 0 | 0 | 0 |
| d1 | 4 | 4 | 6 | 6 |
| d2 | 8 | 8 | 12 | 12 |
| d3 | 12 | 12 | 8 | (18)* |
| d4 | 6 | 16 | 4 | (24)* |
| d5 | 0 | 20 | 0 | (20)* |
| d6 | 4 | 24 | 6 | (16)* |
| d7 | 8 | 18 | 12 | 12 |
| d8 | 12 | 12 | 8 | 8 |
| d9 | 6 | 4 | 4 | 4 |
| d10 | 0 | 0 | 0 | 0 |
Thus, the crystal field splitting depends on the field produced by the ligand and the charge on the metal ion. An experimentally determined series based on the absorption of light by coordination compound with different ligands known as spectrochemical series has been proposed. Spectrochemical series arranges ligands in order of their field strength as:
I– < Br– < Cl– < SCN– < F– < OH– < C2O42- < H2O < NCS– < EDTA4- < NH3 < en < CN– < CO
Filling of d-orbitals takes place in the following manner; the first three electrons are arranged in t2g level as per Hund’s rule. The fourth electron can either enter into t2g level giving a configuration of t2g4eg0 or can enter the eg orbital giving a configuration of t2g3eg1. This depends on two parameters magnitude of crystal field splitting, Δo and pairing energy, P. The possibilities of two cases can better be explained as:
(i) Δo > P: Electron enters in the t2g level giving a configuration of t2g4eg0. Ligands producing this configuration are known as strong field ligands and form low spin complexes.
(ii) Δo < P: Electron enters in the eg level giving a configuration of t2g3eg1. Ligands producing this configuration are known as weak field ligands and form high spin complexes.
Stability of Complexes
A complex is formed in several steps. Each process step is reversible and the equilibrium constant is known as the stepwise formation constant. Let us consider the formation of complex ML4.
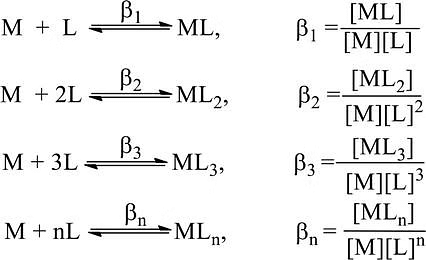
The overall formation constant or stability constant, β = K1 × K2 × K3 × K4 and 1/β = Instability constant
Factors affecting the stability of complex compounds
(i) The values of stability constant differ widely depending on the nature of the metal ion and the ligand In general higher the charge density on the central ion. The greater the stability of its complexes.
(ii) the more basic a ligands, the greater is the ease with which it can donate its lone pairs of electrons and therefore, greater is the stability of the complexes formed by it.
Example: The cyano and ammine complexes are far more stable than those formed by halide ions. This is due to the fact that NH3 and CN- are strong Lewis bases.
(iii) The higher the oxidation state of the metal, the more stable is the complex. The charge density of Co3+ ion is more than Co2+ ion and thus, [Co(NH3)6]3+ is more stable than [Co(NH3)6]2+. Similarly, [Fe(CN)6]3- is more stable than [Fe(CN)6]4-.
(iv) Chelating ligands form more stable complexes as compared to monodentate ligands.
Colour of Complexes
Complexes in which central transition metal ion contains unpaired electrons show colour. It is ‘d – d’ transition.
The colour of complexes depends upon:
- Number of unpaired electrons in transition metal ion
- Nature of ligands
- The oxidation state of central transition metal ion
- The wavelength of light absorbed and emitted
- The proportion of ligands in the coordination sphere
Example: [Ni(H2O)6]+2+ en(aq) → [Ni(H2O)4en]+2 (Green Pale blue)
Bonding in Metal Complexes
Complexes in which carbon monoxide acts as ligands are metal carbonyls
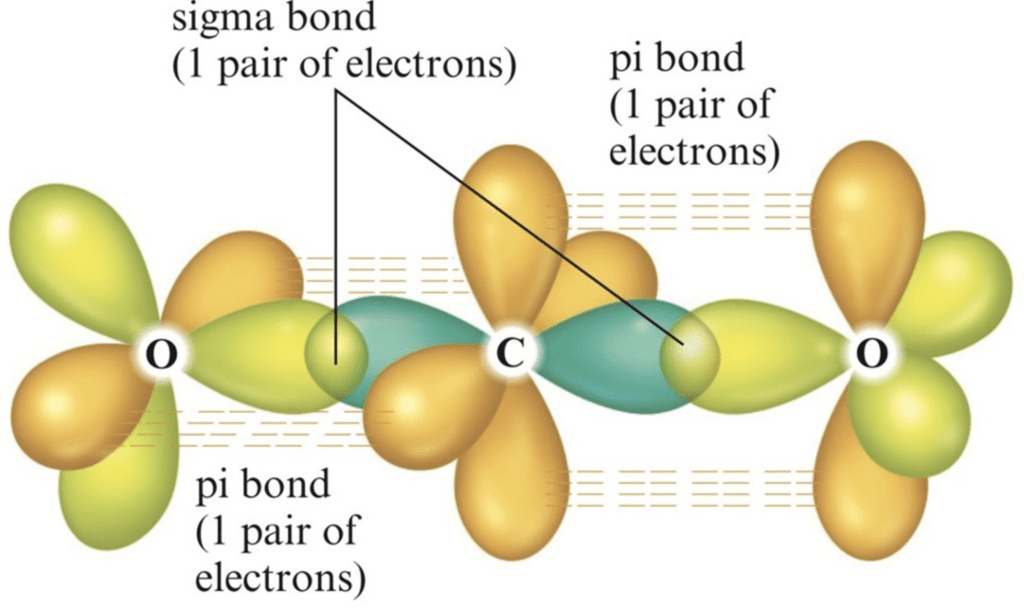
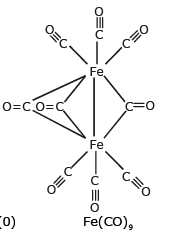
Example: [Ni(CO)4] Tetracarbonyl Nickel (0) and [Fe(CO)5] Penta Carbonyl Iron (0) In these complexes, complexes, a′σ‘ bond is formed by the overlapping of vacant ‘d’ orbital of metal ion and filled orbital of C-atom (carbon).
A π bond is formed by the lateral overlapping of filled inner orbitals of metal ion and vacant of the carbon atom. Thus synergic bonding exists in metal carbonyls.
Applications of Coordination Compounds
The complexes are of immense importance on account of their applications in various fields. During complex formation there are drastic changes in the properties of metal atom/ion these changes in properties are made use of in the application of metal complexes.
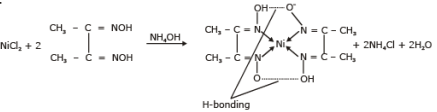
(i) The detection and estimation of Ni2+ is based on the formation of a scarlet red complex with dimethyl glyoxime.
(a) Fe3+ is detected by formation of a blood red coloured complex with KSCN.
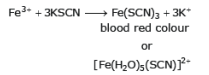
(b) Many ligands (organic reagents) are used for the gravimetric estimation of number of metal ions.
Metal ion to be estimated | Cu2+ | Ni2+ | Fe3+ | Al3+ | Co2+ |
Organic reagents used | Benzoin oxime | Dimethyl glyoxime | 1,20-phena- nthroline | 8-hydroxy quinoline | α-nitroso β-naphthol |
(c) EDTA is used as a complexing agent in volumeter analysis of metal ions like Ca2+ , Mg2+ and Zn2+ .
(d) The co-ordination compounds of the transition metals exhibit a variety of colours. This property is utilised in colorimetric analysis for the estimation of many metals.
(ii) Metallurgical process
Silver and gold are extracted by the use of complex formation. Silver ore is treated with sodium cyanide solution with continuous passing of air through the solution. Silver dissolves as a cyanide complex and silver is precipitated by the addition of scrap zinc.

b) Native Gold and Silver also dissolve in NaCN solution in presence of the oxygen (air).
4 Ag + 8NaCN + O2 + 2H2O  3Na[Ag(CN)2] + 3NaOH
3Na[Ag(CN)2] + 3NaOH
Silver and Gold are precipitated by addition of scrap zinc. Nickel is extracted by converting it into a volatile complex, nickel carbonyl, by use of carbon monoxide (Mond's process). The complex decomposes on heating again into pure nickel and carbon monoxide.
Ni + 4CO  Ni(CO)4
Ni(CO)4 Ni + 4 CO
Ni + 4 CO
(iii) Photography
In photography, the image on the negative is fixed by dissolving all the remaining silver bromide with hypo solution in the form of a soluble complex.
AgBr + 2Na2S2O3 Na3[Ag(S2O3)2 ] + NaBr
Na3[Ag(S2O3)2 ] + NaBr
(soluble) (soluble)
(iv) Electroplating
Metal complexes release metal slowly and give a uniform coating of the metal on the desired object Cyano complexes of silver, gold copper and other metals are used for the electrodeposition of these metals.
(v) Biological processes
Metal complexes are of immense importance in biological processes. Haemoglobin, the red blood pigment, which acts as oxygen carrier to different parts of the body is a complex of iron (II). Vitamin B12 is a complex of cobalt metal. The green colouring matter of plants, called chlorophyll, is a complex of magnesium. It acts as a catalyst in photosynthesis.
Organometallic compounds
Organometallic compounds are defined as those compounds in which the carbon atoms of organic (usually alkyl or aryl) groups are directly bonded to metal atoms. The compounds of elements such as boron, phosphorus, silicon, germanium and antimony with organic groups are also included in organometallics. Many organometallic compounds are important reagents that are used for the synthesis of organic compounds.
Classification of Organometallic Compounds
Organometallic compounds are classified in three classes.
(i) Sigma bonded organometallic compounds: In these complexes, the metal atom and carbon atom of the ligand are joined together with a sigma bond, For Examples:
(a) Grignard reagents, R - Mg - X where R is an alkyl or aryl group and X is a halogen.
(b) Zinc compounds of the formula R2Zn such as (C2H5)2Zn. (isolated by Frankland).
Other similar compounds are (CH3)4Sn, (C2H5)4Pb, Al2(CH3)6, Al2(C2H5)6, Pb(CH3)4 etc.
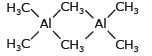
Al2(CH3)6 is a dimeric compound and has a structure similar to diborane, (B2H6). It is an electron-deficient compound and two methyl groups act as bridges between two aluminium atoms.
(ii) Pi-bonded organometallic compounds: These are the compounds of metals with alkenes, alkynes, benzene and other ring compounds. In these complexes, the metal and ligand form a bond that involves the p-electrons of the ligand. Three common examples are Zeise's salt, ferrocene and dibenzene chromium.
These are shown below:
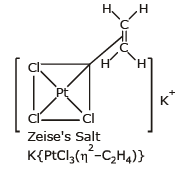
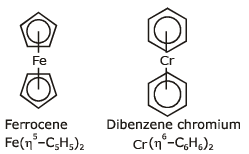
The number of carbon atoms bonded to the metal in these compounds is indicated by the greek letter h(eta) with a number. The prefixes η2, η5 and η6 indicate that 2, 5 and 6 carbon atoms are the metal in the compound.
(iii) Sigma and Pi bonded organometallic compounds: Metal carbonyl compounds formed between metal and carbon monoxide, belong to this class. These compounds possess both s-and p-bonding. Generally, the oxidation state of metal atoms in these compounds is zero. Carbonyls may be mononuclear, bridged or polynuclear.
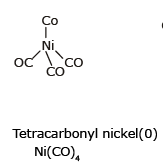
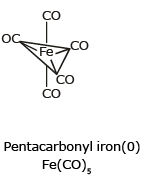
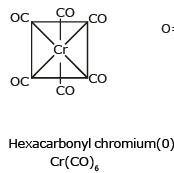
In a metal carbonyl, the metal-carbon bond possesses both the s-and p-character. An s-bond between metal and the carbon atom is formed when a vacant hybrid orbital of the metal atom overlap with an orbital on a C atom of carbon monoxide containing a lone pair of electrons.
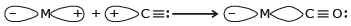
Formation of p-bond is caused when a filled orbital of the metal atom overlaps with a vacant antibonding p* orbital of C atom of carbon monoxide. This overlap is also called back donation of electrons by metal atom to carbon.
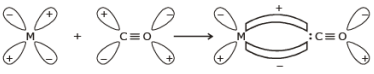
The p-overlap is perpendicular to the nodal plane of the s-bond.
In olefinic complexes, the bonding p-orbital electrons are donated to the empty orbital of the metal atom and at the same time to the back bonding p-orbital of the olefin.
Applications of Organometallic Compounds
(i) Tetraethyl lead (TEL) is used as an antiknock compound in gasoline.
(ii) Wilkinson's catalyst [Rh(PPh3)3Cl] is used as a homogeneous catalyst in the hydrogenation of alkenes.
(iii) The extraction and purification of nickel are based on the formation of organometallic compound Ni(CO)4. The formation of Ni(CO)4 at 50-80ºC and its decomposition at 150-180ºC is used in the extraction of nickel by MOND's Process.
(iv) Zeigler Natta catalyst (trialkyl aluminium titanium tetrachloride) acts as a heterogeneous catalyst in the polymerisation of ethylene into polyethene polymer.
Points to be remembered:
(i) CH3B(OCH3) is an organometallic compound but B(OCH3) is not.
(ii) The closed ring complexes formed by polydentate ligands are called Chelates. Chelation leads to stability.
(iii) Estimation of nickel (II) is done by complexing with dimethyl glyoxime (DMG) whereas that of Ca 2 and Mg2 ions is done by titrating against EDTA.
(iv) Complex in which ligands can be substituted by other ligands is called labile complexes. For example [Cu(NH3)4]2 is a labile complex because NH3 ligands can be substituted by CN- ligands.
[Cu(NH3)4]2+ + 4CN- → [Cu(CN)4]2 + 4NH3
(less stable) (more stable)
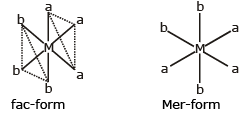
(v) Another type of geometrical isomerism is also shown by octahedral complexes of the type Ma3b3.
if each trio of donor atoms occupy adjacent positions at the corner of an octahedral face, then it is called facial (fac) isomer and when the position are around the meridian of the octahedron, then it is called meridional (mer) isomer.
(vi) Haemoglobin is a complex of Fe, chlorophyll is a complex of Mg, vitamin B12 is a complex of Co.
(vii) s-bond organometallic compounds generally contain a non-transition metal linked to a carbon atom of an alkyl group by s bond. For example eg. R-MgX.
(viii) p-bonded organometallics are formed by the donation of p-electrons of the double bond to the metal atom. For example Zeise's salt K[PtCl3h2 C2H4] and Ferrocene Fe(h5-C5H5)2
(ix) Grignard's reagent is one of the most useful organometallic compounds. Due to the high polarity of (Cd-Mgd ) bond, it can be used to synthesise many organic compounds.
|
75 videos|278 docs|78 tests
|
FAQs on Bonding in Coordination Compounds: VBT and CFT - Chemistry Class 12 - NEET
| 1. What is the history of Valence Bond Theory? |  |
| 2. What are the postulates of Valence Bond Theory? | |
| 3. How many orbitals are involved in hybridization according to Valence Bond Theory? | |
| 4. What are the limitations of Valence Bond Theory? | |
| 5. How does Crystal Field Theory explain the bonding in coordination compounds? | |




study material
,shortcuts and tricks
,past year papers
,Objective type Questions
,Extra Questions
,MCQs
,Bonding in Coordination Compounds: VBT and CFT | Chemistry Class 12 - NEET
,Important questions
,Bonding in Coordination Compounds: VBT and CFT | Chemistry Class 12 - NEET
,Free
,Sample Paper
,Summary
,Exam
,Previous Year Questions with Solutions
,Semester Notes
,Bonding in Coordination Compounds: VBT and CFT | Chemistry Class 12 - NEET
,practice quizzes
,ppt
,mock tests for examination
,Viva Questions
,video lectures
;


Bonding in Coordination Compounds: VBT and CFT Free PDF Download
Importance of Bonding in Coordination Compounds: VBT and CFT
Bonding in Coordination Compounds: VBT and CFT Notes
Bonding in Coordination Compounds: VBT and CFT NEET Questions
Study Bonding in Coordination Compounds: VBT and CFT on the App
|
© EduRev
|
Education Revolution
|



|





