NEET PG Exam > NEET PG Tests > Test: Chemistry and Metabolism of Amino Acids- 3 - NEET PG MCQ
Test: Chemistry and Metabolism of Amino Acids- 3 - NEET PG MCQ
Test Description
25 Questions MCQ Test - Test: Chemistry and Metabolism of Amino Acids- 3
Test: Chemistry and Metabolism of Amino Acids- 3 for NEET PG 2025 is part of NEET PG preparation. The Test: Chemistry and Metabolism of Amino Acids- 3 questions and answers have been prepared
according to the NEET PG exam syllabus.The Test: Chemistry and Metabolism of Amino Acids- 3 MCQs are made for NEET PG 2025 Exam.
Find important definitions, questions, notes, meanings, examples, exercises, MCQs and online tests for Test: Chemistry and Metabolism of Amino Acids- 3 below.
Solutions of Test: Chemistry and Metabolism of Amino Acids- 3 questions in English are available as part of our course for NEET PG & Test: Chemistry and Metabolism of Amino Acids- 3 solutions in
Hindi for NEET PG course.
Download more important topics, notes, lectures and mock test series for NEET PG Exam by signing up for free. Attempt Test: Chemistry and Metabolism of Amino Acids- 3 | 25 questions in 25 minutes | Mock test for NEET PG preparation | Free important questions MCQ to study for NEET PG Exam | Download free PDF with solutions
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 1

Which is elevated in PLP deficiency?
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 1
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 2

Test: Chemistry and Metabolism of Amino Acids- 3 - Question 3
In Phenylketonuria the main aim of first line therapy is: (AIIMS Nov 2010)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 3
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 4
A 40-year-old woman presents with progressive palmoplantar pigmentation X-ray spine shows calcification of IV disk. On adding benedicts reagent to urine, it gives greenish brown precipitate and blue-black supernatant fluid. What is the diagnosis? (AIIMS Nov 2008)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 4
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 5
Dopamine hydroxylase catalyse: (Ker 2007)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 5
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 6

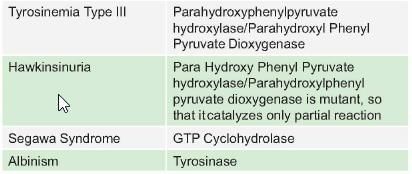
*Multiple options can be correct
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 7
Terminal product of Phenylalanine metabolism is: (PGI May 2014)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 7
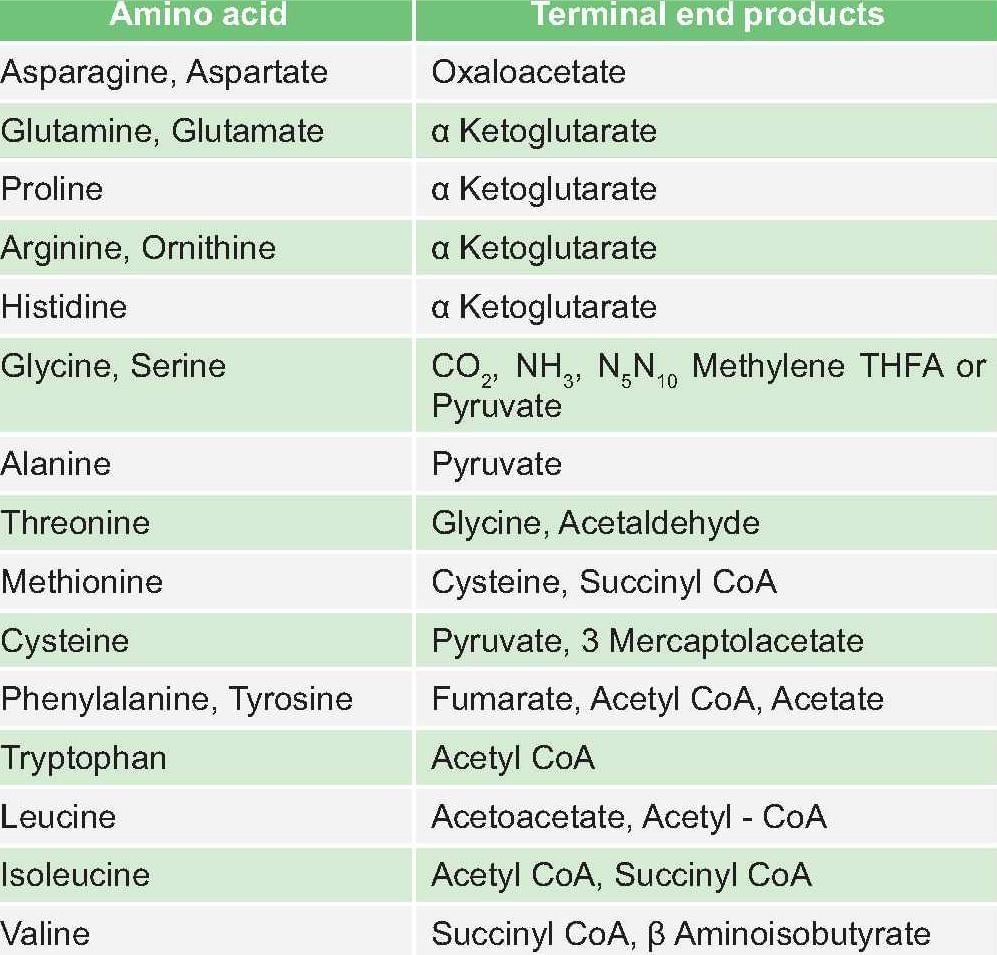
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 8
Enzyme deficiency in albinism is:
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 8
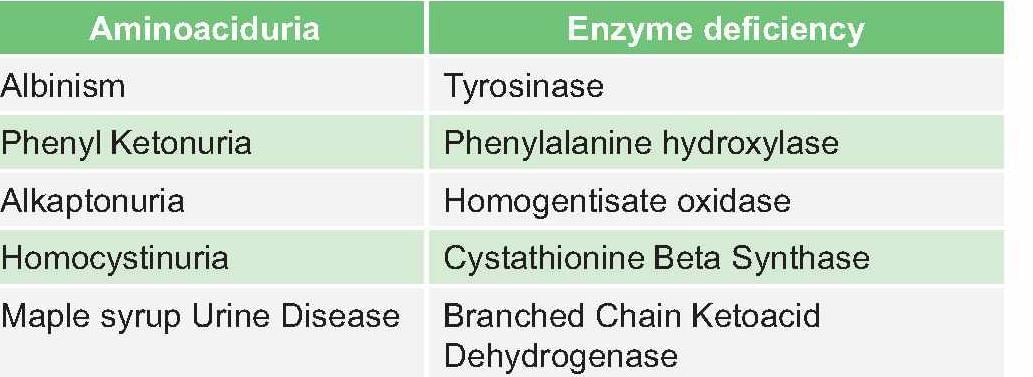
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 9
Mousy body odor is due to: (JIPMER May 2015)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 9
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 10
The amino acid that can be converted into a vitamin: (Kerala 91)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 10
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 11
Which of the following amino acids is involved in the synthesis of thyroxine? (Kamat 97)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 11
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 12
Tyrosinemics are more susceptible to develop:
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 12
*Multiple options can be correct
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 13
Metabolites of tryptophan can give rise to:
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 13
*Multiple options can be correct
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 14
Correct combination of Urine odor in various metabolic disorder: (PGI Nov 2013)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 14
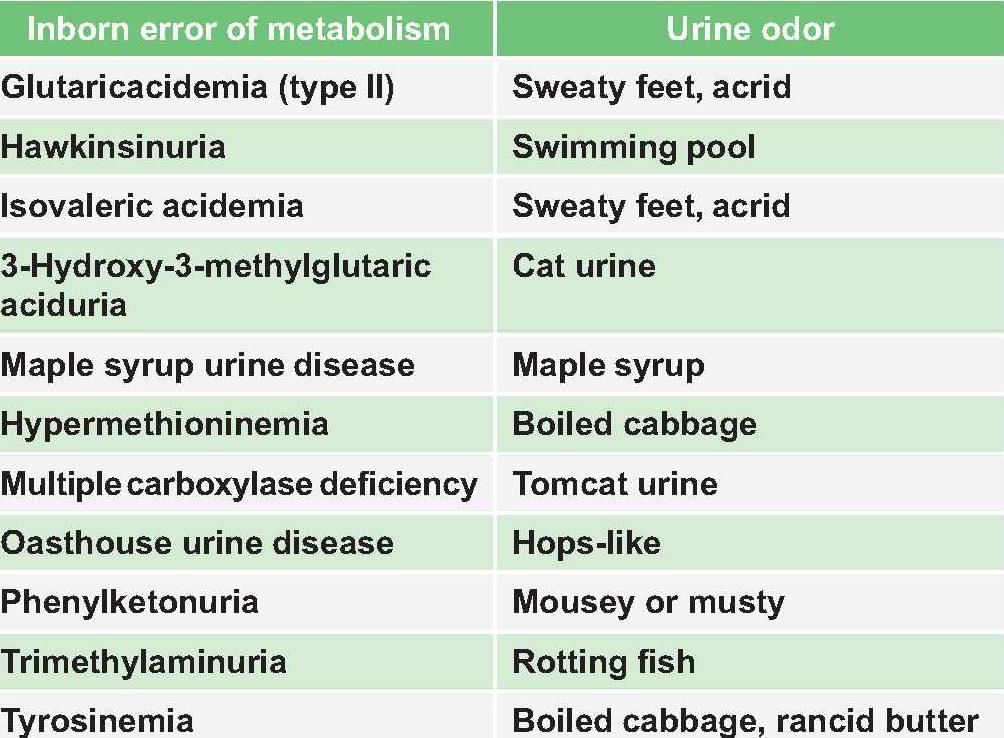
*Multiple options can be correct
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 15
Which of the following is true regarding Phenyl Ketonuria? (PGI nov 2014)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 15
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 16
Which of the following is true about glycine?
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 16
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 17
Which of the following would not act as source of glycine by transamination?
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 17
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 18
Glycine cleavage system in liver mitochondria is associated with which enzyme?
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 18
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 19
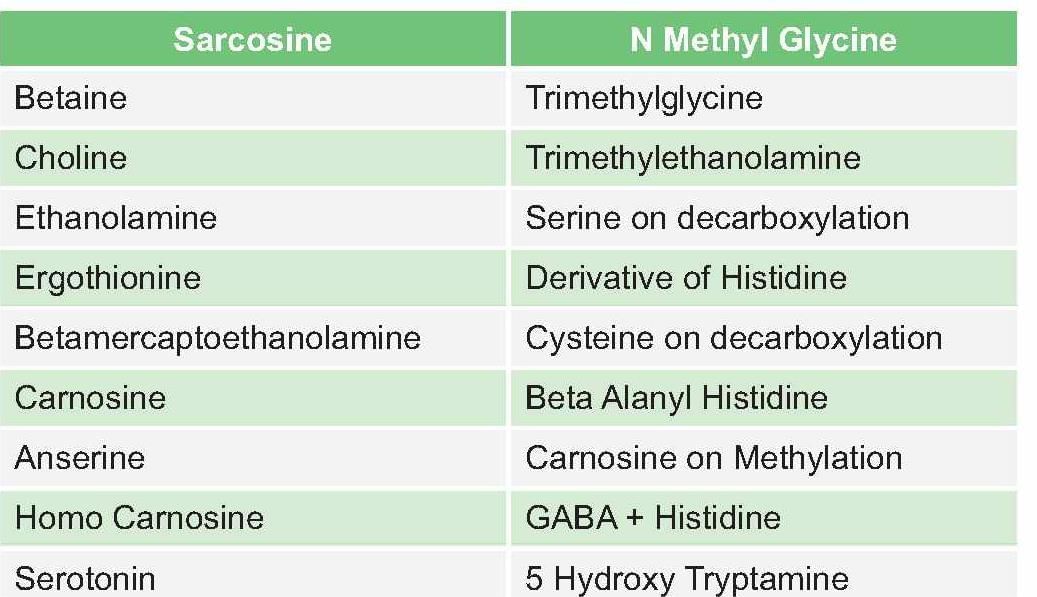
Guanidoacetic acid is formed in from .
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 19
*Multiple options can be correct
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 20
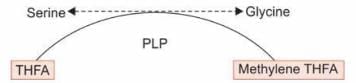
Conversion of glycine to serine requires:
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 20
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 21
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 22
What is the metabolic defect in Primary Oxaluria Type II?
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 22
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 23
All are true about glutathione except:
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 23
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 24
Sulfur of cysteine are not used/utilized in the body for the following process/product: (PGI May 2015)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 24
Test: Chemistry and Metabolism of Amino Acids- 3 - Question 25
Cysteine is abundantly found in: (Ker 2008)
Detailed Solution for Test: Chemistry and Metabolism of Amino Acids- 3 - Question 25
Information about Test: Chemistry and Metabolism of Amino Acids- 3 Page
In this test you can find the Exam questions for Test: Chemistry and Metabolism of Amino Acids- 3 solved & explained in the simplest way possible.
Besides giving Questions and answers for Test: Chemistry and Metabolism of Amino Acids- 3, EduRev gives you an ample number of Online tests for practice
Download as PDF




Important Questions for Chemistry and Metabolism of Amino Acids- 3
Find all the important questions for Chemistry and Metabolism of Amino Acids- 3 at EduRev.Get fully prepared for Chemistry and Metabolism of Amino Acids- 3 with EduRev's comprehensive question bank and test resources.
Our platform offers a diverse range of question papers covering various topics within the Chemistry and Metabolism of Amino Acids- 3 syllabus.
Whether you need to review specific subjects or assess your overall readiness, EduRev has you covered.
The questions are designed to challenge you and help you gain confidence in tackling the actual exam.
Maximize your chances of success by utilizing EduRev's extensive collection of Chemistry and Metabolism of Amino Acids- 3 resources.
Chemistry and Metabolism of Amino Acids- 3 MCQs with Answers
Prepare for the Chemistry and Metabolism of Amino Acids- 3 within the NEET PG exam with comprehensive MCQs and answers at EduRev.
Our platform offers a wide range of practice papers, question papers, and mock tests to familiarize you with the exam pattern and syllabus.
Access the best books, study materials, and notes curated by toppers to enhance your preparation.
Stay updated with the exam date and receive expert preparation tips and paper analysis.
Visit EduRev's official website today and access a wealth of videos and coaching resources to excel in your exam.
Online Tests for Chemistry and Metabolism of Amino Acids- 3
Practice with a wide array of question papers that follow the exam pattern and syllabus.
Our platform offers a user-friendly interface, allowing you to track your progress and identify areas for improvement.
Access detailed solutions and explanations for each test to enhance your understanding of concepts.
With EduRev's Online Tests, you can build confidence, boost your performance, and ace Chemistry and Metabolism of Amino Acids- 3 with ease.
Join thousands of successful students who have benefited from our trusted online resources.
|
© EduRev
|
Education Revolution
|



|









Signup to see your scores
go up within 7 days!
Access 1000+ FREE Docs, Videos and Tests
Takes less than 10 seconds to signup