Heme Metabolism and Hemoglobins Chapter Notes | Biochemistry - NEET PG PDF Download
| Table of contents |
|
| Structure of Heme |
|
| Biosynthesis of Heme |
|
| Porphyrias |
|
| Heme Catabolism |
|
| Hemoglobin |
|
| Myoglobin |
|
| Hemoglobinopathies |
|

Structure of Heme
Heme is a type of metalloporphyrin.
Porphyrins
- Porphyrins are ring-shaped compounds formed by linking four pyrrole rings with methyne or methenyl bridges (–CH–).
- Porphyrinogens are colorless, whereas porphyrins are colored due to the double bonds in the pyrrole ring and the methenyl bridges, which affect their absorption and fluorescence properties.
- All porphyrins exhibit a characteristic absorption band around 400 nm, known as the Soret band, named after the French physicist Charles Soret.
- Porphyrins are utilized in cancer phototherapy because tumors absorb more porphyrins than normal tissues. This process, called Photodynamic therapy, involves activating porphyrins with an argon laser to harm the tumor.
- Porphyrins can cause photosensitivity.
- A unique feature of porphyrins is their ability to form complexes with metal ions, which bind to the nitrogen atoms in the pyrrole rings.
- Examples of Porphyrins:Iron Porphyrins, such as those found in hemoglobin. Magnesium Porphyrins, like chlorophyll, which aids plants in photosynthesis.
- Side Chains in Natural Porphyrins: In nature, porphyrins have various side chains replacing the eight hydrogen atoms in their structure. These side chains include: M. Methyl, A. Acetate, V. Vinyl, P. Propionate.
- Significant Porphyrins:Uroporphyrin, Coproporphyrin, and Protoporphyrin are the three major porphyrins.
- Water Solubility of Porphyrins:Uroporphyrin is the most water-soluble porphyrin, Protoporphyrin is the least water-soluble, and Coproporphyrin has medium water solubility.
Heme consists of a porphyrin ring attached to an iron ion. The specific porphyrin in heme is Protoporphyrin, and the metal ion is iron, specifically in the ferrous state. Therefore, heme is also referred to as Ferroprotoporphyrin.
Important Heme-containing Proteins
- Hemoglobin: Transports oxygen in the blood.
- Myoglobin: Stores oxygen in muscle tissues.
- Cytochrome c: Plays a role in the electron transport chain.
- Cytochrome P450: Involved in the hydroxylation of xenobiotics.
- Catalase: Breaks down hydrogen peroxide.
- Tryptophan pyrrolase: Oxidizes tryptophan.
Biosynthesis of Heme
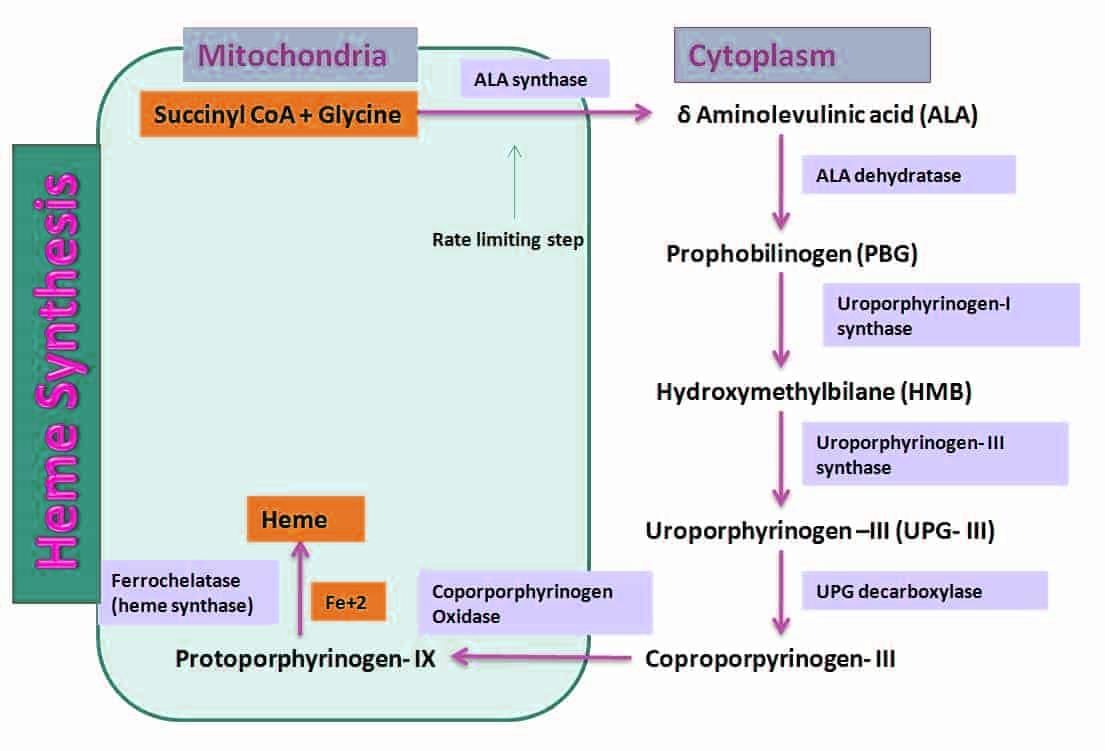
Site of Synthesis: Heme is synthesized in nearly all tissues of the body, except in mature erythrocytes.
- 85% of heme is produced in erythroid precursor cells in the bone marrow.
- The majority of the remaining heme is synthesized in hepatocytes.
Organelle: The synthesis of heme is partly cytoplasmic and partly mitochondrial.
Starting Materials: The process begins with Succinyl CoA and Glycine.
Steps of Synthesis of Heme
The synthesis of heme can be divided into the following steps:
- Synthesis of Porphobilinogen (Monopyrrole)
- Synthesis of Uroporphyrinogen (Tetrapyrrole)
- Conversion of Uroporphyrinogen to Protoporphyrin
- Formation of Heme by incorporating iron into protoporphyrin.
ALA Synthase (ALAS): This enzyme catalyzes the condensation reaction between succinyl-CoA and glycine to form α-amino-β-ketoadipic acid, which is then quickly decarboxylated to produce δ-aminolevulinate (ALA).
The synthesis of ALA occurs in the mitochondria.
Two Isoforms of ALAS
- ALAS-I is distributed throughout the body and plays a crucial role in heme synthesis.
- ALAS-II is specific to cells that produce red blood cells, such as erythroid progenitor cells in the bone marrow.
- ALAS-1 is regulated by heme, which acts as a negative feedback regulator. Heme inhibits ALAS-1 activity.
- ALAS-1 can be increased by certain drugs that require Cytochromes for their metabolism, leading to enhanced heme synthesis.
- ALAS-2 is not subject to heme feedback regulation and is not activated by drugs. Its activity is constant and not influenced by heme levels.
ALA Dehydratase
- ALA dehydratase is an enzyme that plays a crucial role in the biosynthesis of porphyrins, which are important for the production of heme.
- This enzyme catalyzes the condensation of two molecules of 5-aminolevulinic acid (ALA) to produce one molecule of porphobilinogen (PBG), along with the release of two molecules of water.
- The reaction takes place in the cytosol of the cell and involves a zinc ion as a cofactor.
- ALA dehydratase is sensitive to inhibition by lead, which can occur in cases of lead poisoning. Lead interferes with the enzyme's activity by disrupting the zinc cofactor, leading to a decrease in porphyrin synthesis.
- The synthesis of uroporphyrinogen, a cyclic tetrapyrrole, involves the condensation of four molecules of PBG to form a porphyrin. This step is crucial in the production of porphyrin precursors, which are later modified to form heme.
Uroporphyrinogen–I Synthase or HMB Synthase or PBG Deaminase
- Four molecules of PBG join together in a head-to-tail fashion to create a linear tetrapyrrole known as hydroxy-methylbilane ( HMB ).
- During this process, 4 molecules of NH3 are released.
- This reaction occurs in the cytosol.
- The enzyme responsible for this reaction is uroporphyrinogen I synthase, also called PBG deaminase or HMB synthase.
Uroporphyrinogen III Synthase
- HMB is transformed into uroporphyrinogen III by the enzyme uroporphyrinogen III synthase.
- Uroporphyrinogen is the first porphyrin precursor formed.
- Under normal conditions, the uroporphyrinogen produced is almost entirely the III isomer.
- However, in certain porphyrias, HMB may spontaneously cyclise to form uroporphyrinogen I.
Conversion of Uroporphyrinogen to Protoporphyrin by Uroporphyrinogen Decarboxylase
- Uroporphyrinogen III is converted to coproporphyrinogen III through the decarboxylation of all acetate groups, converting them to methyl groups.
- This reaction is catalysed by uroporphyrinogen decarboxylase, which can also convert uroporphyrinogen I to coproporphyrinogen I.
- This process occurs in the cytosol.
- Coproporphyrinogen III then moves into the mitochondria, where it is changed into protoporphyrinogen III.
- The mitochondrial enzyme coproporphyrinogen oxidase catalyses the decarboxylation and oxidation of two propionic side chains to form protoporphyrinogen.
- This enzyme works specifically on type-III coproporphyrinogen, explaining why type I protoporphyrins are not commonly found in nature.
Protoporphyrinogen Oxidase
- The conversion of protoporphyrinogen to protoporphyrin is catalysed by the mitochondrial enzyme protoporphyrinogen oxidase.
Formation of Heme by Incorporation of Iron
- This is the final step in heme synthesis.
- It involves adding ferrous iron to protoporphyrin in a chemical reaction.
- This step is catalysed by ferrochelatase (also known as heme synthase).
- This process takes place in the mitochondria.
Regulation of Heme Synthesis
- ALA synthase is the main regulatory enzyme in the liver's production of heme.
- There are two forms of ALA synthase: hepatic (ALAS1) and erythroid (ALAS2).
- The rate-limiting reaction for heme synthesis in the liver involves ALAS1.
- Heme likely acts as a negative regulator of ALAS1 synthesis, affecting it through a mechanism of repression and derepression.
- The synthesis rate of ALAS1 increases significantly when heme is absent and decreases when heme is present.
Factors that Affect Heme Synthesis
- Drugs that induce hepatic cytochromes, such as barbiturates and griseofulvin.
- Lead inhibits the steps catalysed by ALA dehydratase and ferrochelatase.
- INH reduces the availability of PLP.
- This is why glucose is given to relieve acute porphyrin attacks.
- High levels of glucose in cells stop the induction of ALA synthase.
Porphyrias
Porphyrias are a group of disorders that arise from issues in the body's production of heme, which can be due to either genetic or acquired factors. Most porphyrias are inherited in an autosomal dominant pattern, with a few exceptions.
Exceptions to Autosomal Dominant Inheritance.
- ALA Dehydratase Deficiency Porphyria (ADP)
- Congenital Erythropoietic Porphyria (CEP)
- Erythropoietic Protoporphyria (EPP)
- X-Linked Protoporphyria (XLP)
Clinical Features of Porphyria
Classification of Porphyrias:
1. Erythropoietic Porphyrias:
- Characterized by cutaneous photosensitivity.
- Present at birth or in early childhood.
- Congenital Erythropoietic Porphyria (CEP) may manifest as nonimmune hydrops fetalis during pregnancy.
2. Hepatic Porphyrias:
- Typically appear in adults.
- Porphyria Cutanea Tarda (PCT):
- A type of hepatic porphyria with cutaneous photosensitivity.
- Can occur sporadically.
- Associated with regions where Hepatitis C and HIV are prevalent.
3. Common Types of Porphyria:
- Most common type is Porphyria Cutanea Tarda (PCT).
- Most common acute porphyria is Acute Intermittent Porphyria (AIP).
- In children, the most common porphyria is Erythropoietic Protoporphyria (EPP).
Confirmatory Diagnostic Tests for Porphyrias:
- Enzyme analysis
- Mutation analysis
First-Line Investigations:
- For Porphyrias with Neurovisceral Symptoms: Spot urine ALA and PBG.
- For Porphyrias with Photosensitivity: Plasma porphyrins.
Clinical and Laboratory Features of Various Porphyrias
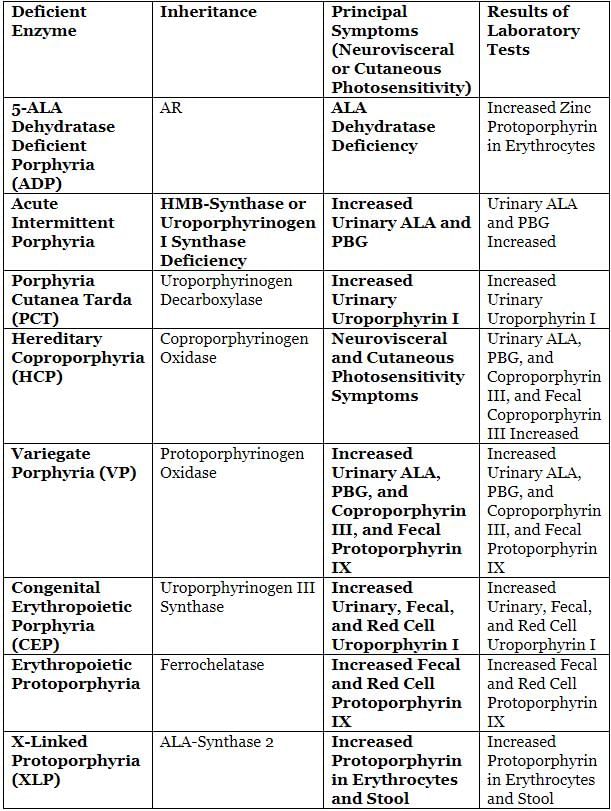
1. ALAD Deficient Porphyria (ADP)
- Also known as Doss Porphyria.
- A rare type of porphyria.
Differential Diagnosis of ADP
- Lead Intoxication: Lead inhibits ALA dehydratase, leading to increased excretion of ALA, similar to ADP.
- Hereditary Tyrosinemia Type I: Succinyl acetone in this condition resembles ALA, causing similarities with ADP.
2. Acute Intermittent Porphyria (AIP)
- Involves a defect in PBG deaminase, HMB synthase, or uroporphyrinogen I synthase.
- The most common acute porphyria.
- Principal symptom is abdominal pain.
- Common physical sign is tachycardia.
- Characterized by neurovisceral symptoms.
- Laboratory findings include increased levels of ALA and PBG, and red-colored urine.
Recent Advances in AIP Treatment
- Liver-directed gene therapy using adeno-associated viral vector (AAV-HMBS) has shown promise in preventing drug-induced AIP.
- Hepatic targeted RNA interference (RNAi) therapy, aimed at reducing high ALAS-1 mRNA levels, has been effective in decreasing ongoing attacks.
Congenital Erythropoietic Porphyria (CEP)
- Also known as Gunther’s Disease.
- Characterized by increased levels of uroporphyrin I in urine, feces, and red blood cells.
Uroporphyrinogen III Synthase Defect
- This condition typically manifests shortly after birth or even in utero as nonimmune hydrops.
- Affected individuals exhibit severe cutaneous photosensitivity.
- Porphyrins accumulate in the teeth and bones of affected individuals.
- This accumulation leads to a brownish discoloration of teeth.
- The defect causes hemolysis due to the presence of erythrocyte porphyrins, resulting in splenomegaly.
- Uroporphyrin I and Coproporphyrin I build up in various parts of the body, including bone marrow, erythrocytes, plasma, urine, and feces.
- Coproporphyrin I is the most commonly found porphyrin in feces.
- Affected individuals may experience port-wine colored urine or pink stains in diapers.
- Erythrodontia is observed, characterized by reddish fluorescence of teeth under long UV light.
- Prenatal diagnosis involves checking porphyrin levels in amniotic fluid and assessing URO synthase enzyme activity in chorionic villi and cultured amniotic cells.
- Beta carotene has the potential to provide protection against sunlight.
- Research is ongoing into gene therapy using human cDNA UROS retroviral vectors.
- Porphyria cutanea tarda (PCT)
- This condition is caused by a defect in uroporphyrinogen decarboxylase.
- Sporadic cases of PCT may be linked to environmental factors and genetic predisposition.
- Aggravating factors for PCT include:
- Infection with Hepatitis C or HIV.
- Excessive alcohol consumption.
- Elevated levels of iron in the body.
- Increased levels of estrogen.
- PCT is the most easily treatable type of porphyria.
- PCT is associated with haemochromatosis.
- The condition leads to blistering skin lesions, primarily on the back of the hands.
- Patients with PCT are at an increased risk for chronic liver disease and hepatocellular carcinoma.
- Treatment options for PCT include:
- Repeated phlebotomy to reduce hepatic iron levels.
- Low-dose treatment with chloroquine or hydroxychloroquine.
- In patients with end-stage renal disease, administration of erythropoietin may be necessary.
- Erythropoietic protoporphyria (EPP)
- EPP results from a defect in ferrochelatase due to mutations in the FECH gene.
- EPP is the most common porphyria in children and the second most common in adults.
- Individuals with EPP experience non-blistering photosensitivity, characterized by pain, swelling, and redness shortly after sun exposure, resembling angioedema.
- Vesicular lesions are rare in EPP.
- A significant increase in erythrocyte protoporphyrin, which is typically free and not bound to zinc, is a hallmark of EPP.
- Erythrocytes in EPP patients exhibit red fluorescence under fluorescence microscopy at 620 nm.
- Genetic testing for FECH mutations can be performed to diagnose EPP.
- Oral beta carotene may help enhance tolerance to sunlight in affected individuals.
- Afamelanotide, an alpha melanocyte-stimulating hormone, is currently undergoing phase III trials for patients with EPP and X-linked protoporphyria.
- X-linked protoporphyria (XLP)
- XLP is caused by increased activity of ALA synthase-2 due to a gain-of-function mutation.
- X-linked sideroblastic anaemia is not classified as a porphyria.
- This condition is caused by decreased activity of ALA synthase-2.
Heme Catabolism
Under normal circumstances in adult humans, approximately 200 billion red blood cells are destroyed each day. A person weighing 70 kg typically breaks down around 6 grams of hemoglobin daily. Each gram of hemoglobin results in the production of about 35 milligrams of bilirubin. Therefore, the daily production of bilirubin in adults ranges from 250 to 350 milligrams.
Fate of Hemoglobin
When hemoglobin is broken down in the body:
- Globin is broken down into its amino acids, which are reused.
- The iron from heme enters the iron pool for reuse.
- The iron-free porphyrin part of heme is mainly broken down in the reticuloendothelial cells of the liver, spleen, and bone marrow.
Steps of Heme Catabolism
Site: Microsomal fraction of reticuloendothelial cells of the liver, spleen, and bone marrow.
Hemoxygenase:
- Oxygen is added to the α-methyne bridge between pyrroles I and II of the porphyrin.
- The tetrapyrrole ring is split.
- Biliverdin, a green pigment, is produced.
- Carbon monoxide is generated by this reaction, which is not the only source of endogenous CO in the body.
Biliverdin Reductase
- This reduces the methyne bridge between pyrrole III and pyrrole IV of biliverdin to a methylene group.
- Bilirubin, a yellow pigment, is produced.
- This process occurs in the cytosol.
Transport of Bilirubin
- Bilirubin formed in peripheral tissues is transported to the liver by plasma albumin.
- Bilirubin is not very soluble in water, but its solubility in plasma increases by noncovalent binding to albumin.
- Each albumin molecule has one high-affinity site and one low-affinity site for bilirubin.
- In 100 ml of plasma, about 25 mg of bilirubin can bind tightly to albumin at the high-affinity site.
- Excess bilirubin can only bind loosely and may easily detach and diffuse into tissues.
Metabolism of Bilirubin
Occurs primarily in the liver and can be divided into three main processes:
- Uptake of bilirubin by liver parenchymal cells.
- Conjugation of bilirubin with glucuronate in the endoplasmic reticulum.
- Secretion of conjugated bilirubin into the bile.
Uptake of Bilirubin in the Liver
In the liver, bilirubin is removed from albumin:
- It is taken up at the sinusoidal surface of hepatocytes by a carrier-mediated system.
- This system has a large capacity and does not limit bilirubin metabolism even in pathological conditions.
- Once inside hepatocytes, bilirubin can bind to certain cytosolic proteins, known as intracellular binding.
Functions of Intracellular Binding
- These proteins may prevent bilirubin from returning to the bloodstream.
- They help to keep bilirubin soluble before conjugation.
Conjugation of Bilirubin with Glucuronic Acid Occurs in the Liver
Hepatocytes convert bilirubin into a polar form by adding glucuronic acid:
- This process, called conjugation, can involve other polar molecules.
- The enzyme that catalyzes this is referred to as bilirubin-UGT.
- Bilirubin monoglucuronide is an intermediate, which is then converted to bilirubin diglucuronide.
- Most bilirubin excreted in bile is in the form of bilirubin diglucuronide.
- However, when bilirubin conjugates are abnormal in plasma (e.g., in obstructive jaundice), they are mostly monoglucuronides.
Transport Mechanisms
- Bilirubin-UGT activity can be induced by various clinically useful drugs, including phenobarbital.
- This process involves an active transport mechanism and is the rate-limiting step for hepatic bilirubin metabolism.
- The protein involved is MRP-2 (multidrug-resistance-like protein 2), located in the biliary canalicular membrane.
- A portion of bilirubin diglucuronide is also transported into portal circulation by MRP-3.
- This is subject to reuptake into hepatocytes by transporters OATP1B1 and OATP1B3.
- The hepatic transport of conjugated bilirubin into bile can be induced by the same drugs that promote bilirubin conjugation.
Fates of Urobilinogen
- In the intestines, bacteria convert conjugated bilirubin into urobilinogen .
- Urobilinogen then travels to the terminal ileum and the large intestine.
- In the intestines, specific bacterial enzymes called β-glucuronidases remove glucuronides from bilirubin.
- This process transforms bilirubin into a group of colourless compounds known as urobilinogens .
Fate of Urobilinogen
- 80-90% of urobilinogen is converted into stercobilinogen and stercobilin by intestinal bacteria and is eliminated in the feces.
- 10-20% of urobilinogen enters the enterohepatic circulation and is returned to the liver.
- A small amount of urobilinogen, less than 3 mg/dL, avoids hepatic uptake, is filtered through the renal glomerulus, and is excreted in the urine.
Hyperbilirubinemias
Jaundice
- Scleral icterus occurs when serum bilirubin levels exceed 3 mg/dL.
- Carotenoderma can be distinguished from icterus by the lack of scleral involvement.
Congenital Hyperbilirubinemias
Unconjugated Hyperbilirubinemias
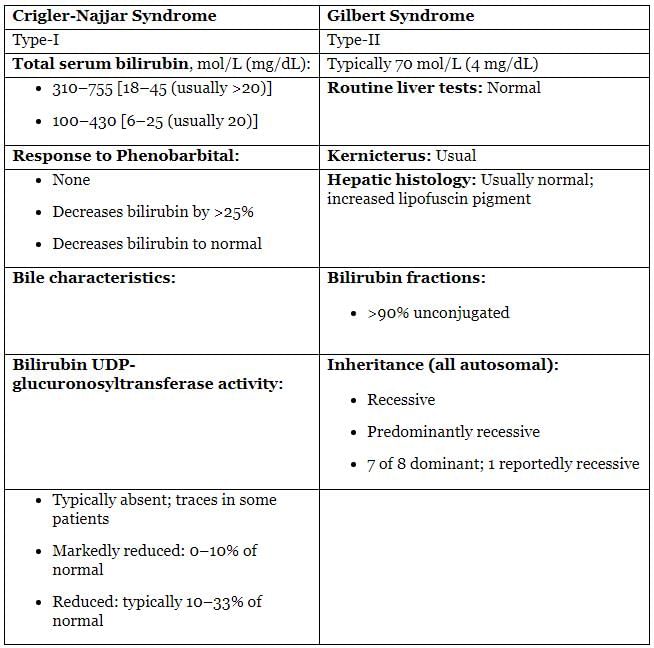
- Gilbert’s disease
- Crigler-Najjar syndrome
Principal Differential Characteristics of Crigler-Najjar and Gilbert syndrome
Physiological Jaundice
- Predominantly unconjugated hyperbilirubinemia is observed.
- Causes include:
- Incompletely developed hepatic system
- Low UDP Glucuronyl Transferase enzyme
- Unconjugated hyperbilirubinemia that develops from the 2nd to 5th day of birth
- Peak level of serum bilirubin is 5–10 mg/dL
- Normal values are reached within 2 weeks.
- Breast milk jaundice occurs when bilirubin levels rise due to substances in breast milk and typically resolves without treatment.
- Bilirubin conjugation is inhibited by certain fatty acids found in breast milk but not in serum.
- A correlation exists between the epidermal growth factor content in breast milk and elevated bilirubin levels in these infants.
Lucey Driscoll Syndrome
- Transient familial neonatal hyperbilirubinemia.
- UGT1A1 inhibitor is in maternal serum, not in breast milk unlike breast milk jaundice.
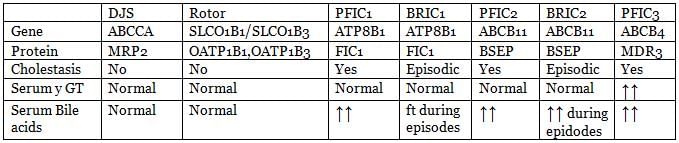
Conjugated Hyperbilirubinemias
- Dubin Johnson’s syndrome
- Rotor syndrome
- Benign Recurrent Intrahepatic Cholestasis (BRIC)
- Progressive Familial Intrahepatic Cholestasis (FIC)
Dubin Johnson’s Syndrome
- Inheritance: Autosomal recessive
- Genetic Cause: Mutation of the gene encoding MRP2 (Multidrug Resistance-Associated Protein 2).
- Liver Histology: Buildup of dark, coarse pigment in the lysosomes of centrilobular hepatocytes, giving the liver a characteristic black appearance. This pigment is thought to be a bilirubin metabolite that is not excreted properly.
- Clinical Features: Black liver jaundice, where the liver's dark appearance is a key feature.
- Diagnostic Test: Brom Sulphthalein test (BSP test) shows 2 peaks, indicating impaired bilirubin excretion.
Rotor Syndrome
- Mechanism: Defective bilirubin excretion due to recent findings suggesting deficiency in plasma transporters OATP1B1 and OATP1B3.
- Pathophysiology: Deficiency in these transporters leads to reduced reuptake of conjugated bilirubin that is pumped into the portal circulation.
- Clinical Features: Jaundice due to accumulation of conjugated bilirubin in the plasma.
Benign Recurrent Intrahepatic Cholestasis (BRIC)
- Overview:. rare disorder characterized by recurrent episodes of jaundice and itching.
- Types: BRIC-1 and BRIC-2.
- BRIC-1: Mutation in the FIC-1 gene, which affects biliary canalicular excretion. This type is distinct from BRIC-2 and is associated with different clinical features.
- BRIC-2: Mutation in the Bile Salt Excretory Protein (BSEP), a protein also defective in Familial Intrahepatic Cholestasis type 2 (FIC-type 2). This type is characterized by recurrent cholestasis and is part of a broader spectrum of bile acid transport disorders.
Progressive Familial Intrahepatic Cholestasis (FIC)
- FIC Type 1 (Byler’s disease): Caused by FIC-1 mutation. Symptoms appear in early infancy as cholestasis, initially episodic. Unlike BRIC, Byler’s disease progresses to malnutrition, growth retardation, and end-stage liver disease.
- FIC Type II: Due to mutation of Bile Salt Excretory Protein (BSEP). This type is characterized by progressive cholestasis and liver failure.
- FIC Type III: Due to mutation of MRP-3 (Multidrug Resistance Protein 3). This type also leads to cholestasis and liver disease but is less common than FIC Type I and II.
Differentiating features of important Conjugated Hyperbilirubinemia
Acquired Hyperbilirubinemias
- Hemolytic Jaundice
- Hepatic Jaundice
- Obstructive Jaundice
Laboratory Tests Done to Differentiate Jaundice
- By Van den Bergh test
- Chemical method to estimate bilirubin in serum
- Bilirubin + Ehrlich’s Diazoreagent (Diazotized Sulfanilic Acid)
- Reddish purple azopigment is formed.
- Analyzed by photometry at 540 nm.
- Two types of reaction: Direct and Indirect.
Conjugated Bilirubin
- Conjugated bilirubin is also known as direct bilirubin.
Unconjugated Bilirubin
- Unconjugated bilirubin is referred to as indirect bilirubin.
Tests in Urine
- Urine and fecal urobilinogen are tested using Ehrlich’s test.
- Urine bilirubin (bile pigment) is tested with Modified Fouchet’s test.
- Urine bile salt is tested by Hay’s test and Pettenkofer test.
Liver Enzyme Panel
- Transaminases (AST and ALT) are elevated in hepatitis.
- Alkaline Phosphatase is elevated in obstructive jaundice.
- Elevated 5’ Nucleotidase levels indicate obstructive jaundice.
Laboratory Tests in Different Types of Jaundice
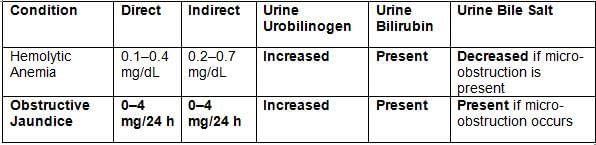
Obstructive Jaundice
- Delta Bilirubin or Biliprotein is a form of conjugated bilirubin that is covalently linked to albumin.
- The half-life of delta bilirubin is approximately 12–14 days, while the half-life of unbound bilirubin is only about 4 hours.
- In cases of conjugated hyperbilirubinemia, bilirubinuria (the presence of bilirubin in urine) occurs later.
Hemoglobin
Hemoglobin is a protein present in red blood cells. It consists of two pairs of chains: a pair of alpha-like chains, each containing 141 amino acids, and a pair of beta-like chains, each with 146 amino acids. Each chain has a heme group, which consists of a protoporphyrin IX ring with an iron atom in the ferrous state (Fe2+). Each heme group can bind to one oxygen molecule, allowing a single hemoglobin molecule to carry up to four oxygen molecules.
Higher-Order Structure of Hemoglobin
- Secondary Structure: The globin chains primarily exhibit an alpha-helix configuration.
- Tertiary Structure: Hemoglobin has a globular shape, with polar amino acids on the surface, enhancing its solubility. The interior contains nonpolar groups, creating a pocket for the heme.
- Quaternary Structure: Hemoglobin is a tetramer made up of two alpha-beta dimers. The tetramer is soluble, but the individual chains are not. If the globin chains are unpaired, they can precipitate and cause cellular damage.
Genetics of Alpha and Beta Chains
Human hemoglobins are produced by two closely linked gene clusters: the α-like genes on chromosome 16 and the β-like genes on chromosome 11.
Types of Hemoglobin
- Embryonic Hemoglobins:
- Hb Portland - ζ2γ2
- Hb Gower I - ζ2ε2
- Hb Gower II - α2ε2
- Fetal Hemoglobin: Hb F - α2γ2
Characteristics of Fetal Hemoglobin
- Slower electrophoretic mobility.
- Greater resistance to alkali denaturation.
- Lesser interaction with 2,3 BPG.
Adult Hemoglobins
- Hb A1 - α1β2
- Hb A2 - α2δ2
Characteristics of Hemoglobin
Hemoglobin is an allosteric protein. The binding of one oxygen molecule to a heme increases the likelihood of oxygen binding at other heme sites, known as positive cooperativity. As a result, the oxygen dissociation curve appears sigmoidal.
Functions of Hemoglobin
The primary functions of hemoglobin include:
- Transporting O2 to body tissues.
- Returning CO2 and protons to the lungs.
Functional Histidine Residues in Hemoglobin
- Proximal Histidine [F8] and Distal Histidine [E7] are key histidine residues.
- The fifth position of iron is occupied by a nitrogen from the imidazole ring of proximal histidine, His F8.
- Distal Histidine, His E7, is located on the opposite side of the heme ring from His F8.
Conformational States of Hemoglobin
- There are two states: T (taut) state and R (relaxed) state.
- The T state has low affinity for oxygen and is the deoxygenated form.
- Transition from T to R state occurs when salt bridges break.
- The R state has high affinity for oxygen and is the oxygenated form.
- The T state promotes oxygen delivery to tissues.
- The R state favours oxygen binding to hemoglobin.
- Binding of the first oxygen molecule shifts the structure from T to R state.
- Binding of 2,3 BPG stabilises the T state, shifting the oxygen dissociation curve to the right.
Interaction of Hemoglobin with 2,3 BPG
- 2,3 BPG is a byproduct produced during glycolysis. It is formed with the assistance of the 2,3 BPG shunt or Rapaport-Leubering cycle. The hemoglobin tetramer binds one molecule of 2,3 BPG in the central cavity formed by the four subunits. 2,3 BPG forms salt bridges with terminal amino groups of both β globin chains through valine, lysine, and histidine.
- This binding reduces hemoglobin's affinity for oxygen by stabilising the T state, shifting the oxygen dissociation curve to the right. 2,3 BPG has a reduced interaction with fetal hemoglobin (HbF). This is because the histidine that forms salt bridges with 2,3 BPG is absent in the γ chain of HbF; instead, there is serine, leading to less interaction and a higher affinity for oxygen in HbF.
Glycated Hemoglobins
- Glycated hemoglobins are formed when glucose or other sugars attach to the alpha and beta chains of hemoglobin A. This nonenzymatic process, known as glycation, is part of the Maillard reaction. Approximately 80% of glucose binds to the N-terminal valine of the beta chain, resulting in HbA1c. The "A" in HbA1c stands for Aldimine, which refers to the linkage between hemoglobin and the NH2 group of valine. HbA1c serves as an important indicator of long-term blood glucose control.
- Since red blood cells have an average lifespan of about 60 days, the level of glycated hemoglobin (HbA1c) reflects average blood glucose levels over the preceding 6 to 8 weeks. The normal range for HbA1c is below 5.5%. A HbA1c level between 5.5% and 6% indicates good blood glucose control.
HbA1c Derived Average Glucose Value
- HbA1c level: Average whole blood glucose:
- 5% - 110 mg/dL
- 6% - 126 mg/dL
- 7% - 154 mg/dL
- 8% - 183 mg/dL
- 9% - 212 mg/dL
- 10% - 240 mg/dL
Calculation of Average Blood Glucose Values from HbA1c
- Average plasma blood glucose (mg/dL) = (HbA1c x 35.6) - 77.3
Myoglobin
Myoglobin is a protein found in muscle tissue. It plays a crucial role in storing and supplying oxygen to muscle cells, particularly during periods of intense exercise when the demand for oxygen increases. Here are some key characteristics of myoglobin:
- Location: Myoglobin is found in muscle tissues, where it helps store and transport oxygen.
- Structure: Myoglobin is a monomer, meaning it consists of a single polypeptide chain. Its molecular weight is approximately 17 kDa.
- Oxygen Storage: Myoglobin stores oxygen in red muscle fibers. During intense exercise, when the demand for ATP (adenosine triphosphate) increases, myoglobin releases this stored oxygen for use in muscle mitochondria to produce ATP.
- Structural Features: Myoglobin is rich in alpha helices, which are a common structural feature in proteins. This structure is important for its function.
- Oxygen Binding: Myoglobin binds only one mole of oxygen at a time. It has a high affinity for oxygen, meaning it can effectively capture and hold onto oxygen molecules.
- Bohr Effect: Myoglobin does not exhibit the Bohr effect, which is a phenomenon where the affinity of hemoglobin for oxygen decreases in the presence of higher levels of carbon dioxide or lower pH.
- Cooperativity: Myoglobin does not display cooperativity in oxygen binding. This means that the binding of oxygen to one myoglobin molecule does not influence the binding of oxygen to another molecule.
- 2, 3 BPG Interaction: Myoglobin does not interact with 2, 3 BPG (2,3-bisphosphoglycerate), a molecule that affects the binding of oxygen to hemoglobin.
- Oxygen Dissociation Curve: The Oxygen Dissociation Curve (ODC) for myoglobin is hyperbolic, indicating a consistent relationship between oxygen saturation and partial pressure of oxygen. This is in contrast to the sigmoidal curve seen in hemoglobin, which reflects its cooperative binding nature.
Hemoglobinopathies
Classification of Hemoglobinopathies
- Name of Hemoglobinopathy
- Altered functional or physical and chemical properties
Structural Hemoglobinopathies
- Structural hemoglobinopathies involve alterations in the amino acid sequences of hemoglobin.
- Examples include:
- HbS (Sickle Hemoglobin). A mutation where glutamic acid (Glu) at position 6 is replaced by valine (Val), leading to abnormal polymerization of hemoglobin.
- HbC. A mutation similar to HbS but with glutamic acid replaced by lysine (Lys) at position 6.
- Altered Oxygen Affinity. Mutations like Hb Yakima (Asp→His at position 99) increase oxygen affinity, causing polycythemia, while others like Hb Kansas (Asn→Lys at position 102) decrease oxygen affinity, leading to cyanosis and pseudoanemia.
- Hemoglobin that Oxidizes Readily. Examples include unstable hemoglobins like Hb Philly (Tyr→Phe at position 35), Hb Genova (Leu→Pro at position 28), and Hb Koln (Val→Met at position 98).
- M Hemoglobins. These involve mutations where proline is replaced by tyrosine, such as in HbM Iwata (His→Tyr at position 87) and HbM Saskatoon, Hyde Park, Milwaukee.
Thalassemias
- Thalassemias are characterized by defective biosynthesis of globin chains.
- α Thalassemias involve defective biosynthesis of α globin chains.
- β Thalassemias involve defective biosynthesis of β globin chains.
- Thalassemic Hemoglobin Variants are structurally abnormal hemoglobins associated with co-inherited thalassemic phenotypes.
- Examples include:
- HbE. A mutation where glutamic acid is replaced by lysine at position 26 of the β globin chain.
- Hb Constant Spring. A mutation in the alpha chain’s C-terminal termination codon, resulting in an elongated alpha chain of 173 amino acids.
- Hb Lepore. A fusion of the proximal end of the δ gene with the distal end of the β gene due to unequal crossover and recombination.
- Hereditary Persistence of Fetal Hemoglobin. This condition involves the persistence of high levels of fetal hemoglobin (HbF) into adult life.
Sickle Cell Anemia
- The molecular defect in sickle cell anemia is caused by a mutation in the β globin chain.
- Specifically, it involves a missense mutation at codon 6, where glutamic acid (GAG) is replaced by valine (GTG), resulting in the substitution of a polar amino acid with a nonpolar one.
- This mutation leads to the pathological changes associated with sickle cell disease.
- When hemoglobin S (HbS) is deoxygenated, it polymerizes reversibly, forming a gelatinous network of fibrous polymers that stiffen the red blood cell (RBC) membrane.
- This process increases blood viscosity and causes dehydration due to potassium leakage and calcium influx.
- The combination of these changes produces the characteristic sickle or honeycomb shape of the RBCs.
- Sickled cells lose their ability to traverse small capillaries and exhibit altered "sticky" membranes, making them abnormally adherent to the endothelium of small venules.
- These abnormalities trigger unpredictable episodes of microvascular vasoocclusion and premature RBC destruction, leading to hemolytic anemia.
Thalassemia Syndromes
- Alpha Thalassemia Syndromes are associated with deletion of α gene loci, as the α chain is encoded by two tightly linked gene clusters.
- Different syndromes are associated with deletion of each gene locus:
- Silent Carrier. 1 α gene locus deleted (-α/αα), HbA1 %: 98–100.
- Thalassemia Trait. 2 α gene loci deleted (-α/-α or --/αα), HbA1 %: 85–95.
- HbH Disease. 3 α gene loci deleted (--/-α), HbA1 %: 70–95, HbH(β4) %: 5–30.
- Hydrops Fetalis. All four loci deleted, NB: 90–95% is Hb Barts (γ4).
Mutations in Alpha and β Thalassemia
- Alpha Thalassemia. Mutations can occur due to unequal crossing-over, large deletions, and less commonly nonsense and frameshift mutations.
- β Thalassemia. Mutations can occur due to deletions, nonsense and frameshift mutations, splice site mutations, and promoter mutations.
- Hb H. This is a β4 tetramer.
- Hb Barts. This is a γ4 tetramer.
Novel Treatments for Hemoglobinopathies
- For Sickle Cell Anemia. Direct gene correction in situ using genomic editing techniques like Zn finger nucleases or CRISPR Cas 9.
- De-repressing HbF by interfering with Bcl 11a.
- For Sickle Cell and Thalassemia Syndromes. Bone marrow transplantation to provide stem cells, used in a large number of β Thalassemia cases and some cases of sickle cell anemia.
- Re-establishing high levels of HbF by stimulating proliferation of primitive Hb F producing progenitor cells using Cytarabine, Hydroxy Urea, or pulsed/intermittent administration of Butyrates.
|
48 docs|7 tests
|
FAQs on Heme Metabolism and Hemoglobins Chapter Notes - Biochemistry - NEET PG
| 1. What is the process of heme synthesis and what are the key steps involved? |  |
| 2. What are porphyrias and how do they relate to heme metabolism? | |
| 3. What are the clinical manifestations of porphyria, and how are they diagnosed? | |
| 4. How does iron incorporation into protoporphyrin IX occur during heme synthesis? | |
| 5. What is the significance of heme in the body, particularly in relation to hemoglobin? | |


Heme Metabolism and Hemoglobins Chapter Notes | Biochemistry - NEET PG
,Exam
,Objective type Questions
,Heme Metabolism and Hemoglobins Chapter Notes | Biochemistry - NEET PG
,Viva Questions
,Sample Paper
,Important questions
,shortcuts and tricks
,Extra Questions
,Free
,video lectures
,MCQs
,Heme Metabolism and Hemoglobins Chapter Notes | Biochemistry - NEET PG
,mock tests for examination
,study material
,Semester Notes
,Previous Year Questions with Solutions
,past year papers
,practice quizzes
,ppt
,Summary
;


Chapter Notes: Heme Metabolism and Hemoglobins Free PDF Download
Importance of Chapter Notes: Heme Metabolism and Hemoglobins
Chapter Notes: Heme Metabolism and Hemoglobins
Chapter Notes: Heme Metabolism and Hemoglobins NEET PG Questions
Study Chapter Notes: Heme Metabolism and Hemoglobins on the App
|
© EduRev
|
Education Revolution
|



|





