Reaction Mechanism Chemistry | Organic Chemistry PDF Download
| Table of contents |
|
| Addition Reactions |
|
| Nucleophilic Addition Reaction |
|
| Substitution Reactions |
|
| Electrophilic (Aromatic) Substitution |
|

In chemistry, a reaction mechanism is the step by step sequence of elementary reactions by which overall chemical change occurs. Organic reactions have been broadly classified into two types on the basis of bond cleavage.
- Free radical reactions (homolytic cleavage)
- Ionic reactions (heterolytic cleavage)
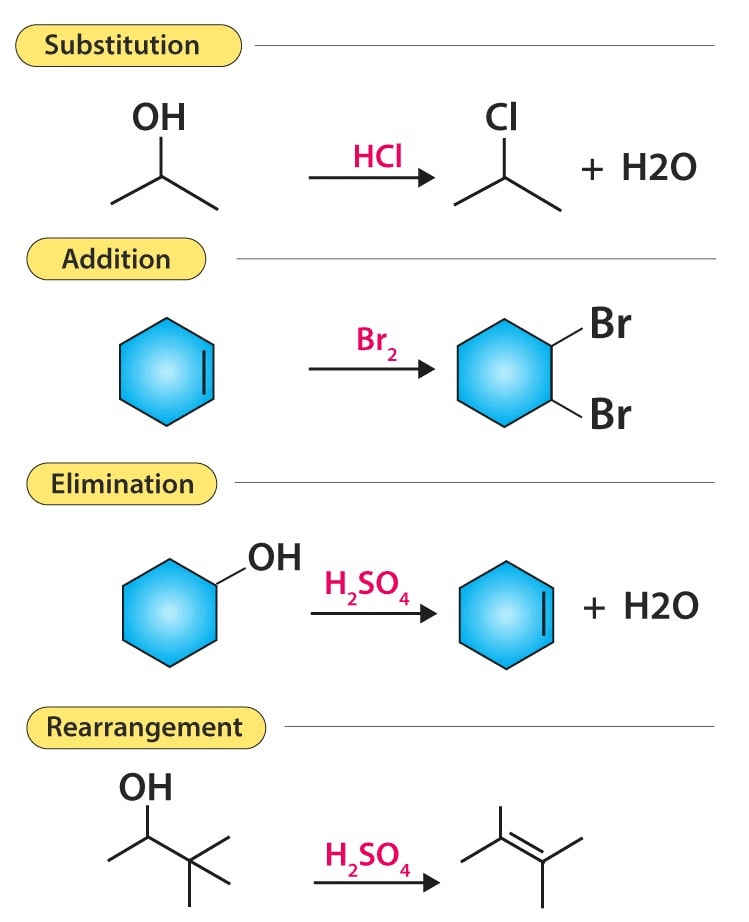
The second type of yet another broad classification is on the basis of the products of the reactions.
- Addition reaction
- Substitution reaction
- Elimination reaction
- Rearrangement
The above two classifications do not specify the reaction type. One satisfactory classification has been made by sub classifications of the latter three types of reactions 1, 2, and 3 on the basis of the reagents involved in the rate-determining step, i.e., the slowest step. Thus,
1. Addition reactions
(i) Electrophilic
(ii) Nucleophilic
2. Substitution reactions
(i) Free radical
(ii) Nucleophilic- SN1, SN2, and SNi
(iii) Electrophilic
3. Elimination reactions
(i) E1
(ii) E2
4. Rearrangement reactions
Addition Reactions
A reaction in which the substrate and the reagent add up to form a product is called an addition reaction.
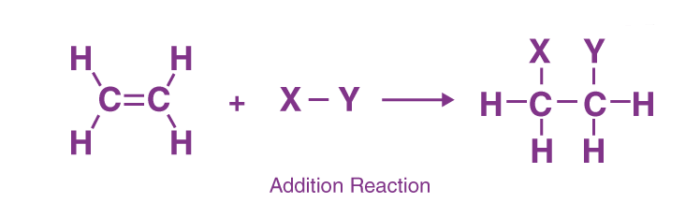
- The reaction occurs at the site of unsaturation in a molecule. Thus, compounds having multiple bonds such as
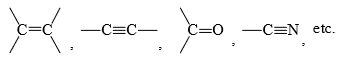
undergo addition reactions. - The reactivity of these compounds is due to the more exposed and easily available π electrons to the electron-seeking (electrophilic) reagent.
(i) Electrophilic Addition Reaction
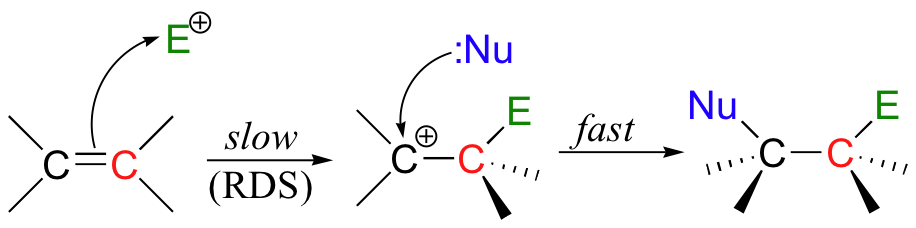
An electrophilic addition reaction is an addition reaction where a chemical compound containing a double or triple bond has a π bond broken, with the formation of two new σ bonds.
This is a characteristic reaction of unsaturated hydrocarbons, e.g.,
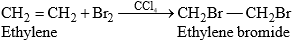
Both the reactants add to form a single product.
Mechanism
The mechanism of the reaction is based on the following observations:
- When ethylene is brominated in an aqueous solution in the presence of sodium chloride, the product in addition to ethylene dibromide contains 1-chloro-2-bromoethane.
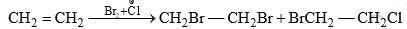
This indicates that a carbocation formed in the first step is attacked by both Br– (from Br2) and Cl– (present in solution) in the second step. - On bromination, trans-2-butene gives meso-dibromide and cis-2-butene gives racemic (DL) modification. This indicates the addition of bromine atoms to the π bond from opposite sides of the plane of the ethylene molecule i.e., trans-addition results.
On the basis of the above observations, the following mechanism is suggested:
- On the approach of the two reactants, the Br—Br bond is polarized by the π electrons to form a π complex which breaks down to form a cyclic bromonium ion and the other bromine atom leaves with the bonding pair of electrons in the first step.
- In the second step the bromide ion attacks either of the carbons (originally double-bonded) from the opposite side to complete the addition. This results in trans-addition.
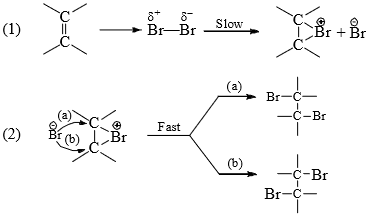
Effect of Substituents
- It is seen that a bromonium ion or a carbocation is formed in the rate-determining step. It is logical to assume that electron-donating substituents will not only increase the electron density for the facile electrophilic attack but also aid to stabilize the carbocation.
- Thus, the activation energy for the formation of the carbocation is lowered so that the reaction becomes fast. The relative rates of addition of some substituted alkenes are.
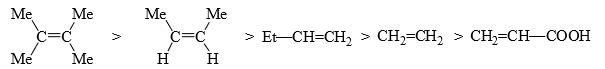
Obviously, electron-withdrawing groups will have the reverse effect.
Markovnikov Rule
- In the case of addition of unsymmetrical reagents (e.g., HX) to unsymmetrical alkenes, the negative moiety (i.e., nucleophile) of the addendum (reagent) attacks the more highly substituted carbon. This is known as the Markovnikov rule.
- Thus, the addition of HBr to propylene gives 2-bromopropane (16) and not 1-bromopropane (17).
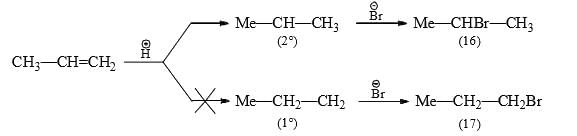
Markovnikov's rule is explained on the basis of the stability of the first-formed carbocation. Since 2° carbocation is more stable and hence formed more easily than 1° carbocation, the product is (16). - Thus, when more than one carbocation formation is possible, the most stable of the carbocations will determine the orientation of the overall addition. This is seen in the addition of HCl to vinyl chloride when ethylidene chloride (18) and not ethylene chloride (19) is obtained.
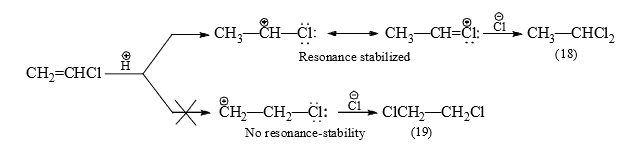
The rate of addition is, however, slower (about 30 times) than with ethane due to the electron-withdrawing effect of chlorine. - In the presence of peroxide or under conditions of radical formation, anti-Markovnikov addition results. The peroxide breaks down to a free-radical which generates a bromine-free radical from HBr. The bromine radical attacks the π electrons producing a 2° radical rather than a 1° radical for reasons of stability.
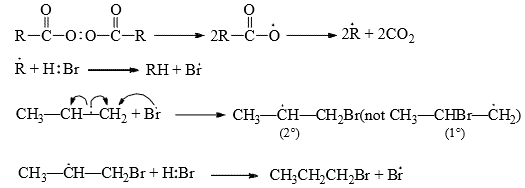
Free radical addition of HF, HI or HCl is energetically unfavorable.
Nucleophilic Addition Reaction
A nucleophilic addition reaction is a chemical addition reaction in which a nucleophile forms a sigma bond with an electron deficient species.
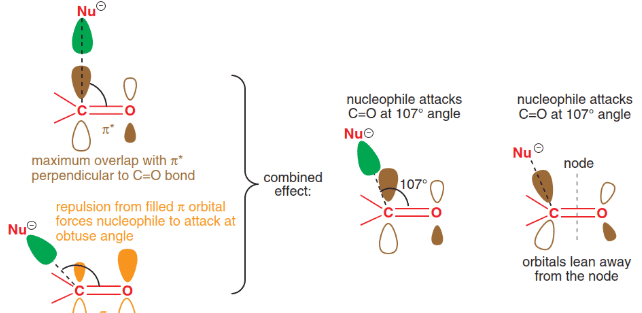 Mechanism of Nucleophilic addition
Mechanism of Nucleophilic addition
- We have seen that electron-releasing groups conjugated to carbon-carbon multiple bonds promote electrophilic addition. In the same way, conjugation of electron-withdrawing groups activates the carbon-carbon multiple bonds towards nucleophilic addition.
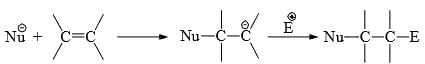
- The substituents reduce the π-electron density, thereby aid the attack of the nucleophile and stabilize the carbanion formed on the attack by delocalization of the negative charge (cf Michael reaction). Some common electron-withdrawing groups are
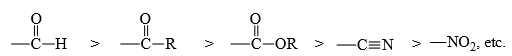
- Polar functional groups, e.g.,
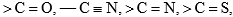 etc., also undergo nucleophilic addition. The hetero atoms, like the electron-withdrawing substituents, reduce the π-electron density on the carbonyl carbon and stabilize the anion, formed on the attack of the nucleophilic, by accommodating the –ve charge.
etc., also undergo nucleophilic addition. The hetero atoms, like the electron-withdrawing substituents, reduce the π-electron density on the carbonyl carbon and stabilize the anion, formed on the attack of the nucleophilic, by accommodating the –ve charge.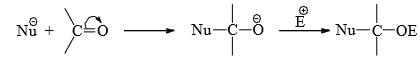
- However, the addition product will be stable enough to be isolated provided the carbonyl group is no attached to a good leaving group such as OH, OR, Cl, NH2, etc. Carboxylic acids and their derivatives, therefore, undergo substitution rather than addition on attack of nucleophiles.
- Nucleophilic addition to carbonyl group is, therefore, a characteristic reaction of aldehydes and ketones since
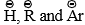 are not good leaving groups. Considering the steric and electronic factors (inductive effect) of the group attached to the carbonyl carbon, the reactivity of the carbonyl groups decreases in the order: H2C=O > RCHO > R2CO > ArCHO > Ar2CO.
are not good leaving groups. Considering the steric and electronic factors (inductive effect) of the group attached to the carbonyl carbon, the reactivity of the carbonyl groups decreases in the order: H2C=O > RCHO > R2CO > ArCHO > Ar2CO.
HCN Addition to Carbonyl Group
- The carbonyl group may be considered as a resonance hybrid of the following canonical structures.
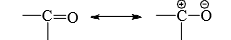
- The addition of the nucleophile
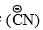 to the carbon will be facilitated by adding a base to increase the concentration of
to the carbon will be facilitated by adding a base to increase the concentration of 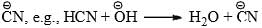 or by adding acid to protonate the oxygen atom so as to increase the +ve character of the carbonyl carbon, e.g.,
or by adding acid to protonate the oxygen atom so as to increase the +ve character of the carbonyl carbon, e.g., 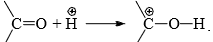 Hence, the reaction is catalyzed by both acids and bases. However, base-catalyzed nucleophilic addition reactions are common.
Hence, the reaction is catalyzed by both acids and bases. However, base-catalyzed nucleophilic addition reactions are common. - Base-catalyzed- This removes the weakly acidic protons from the reagent to generate the nucleophile which adds to the carbonyl carbon in the first step. In the second step, a fast addition of protons to the negatively charged oxygen completes the addition.
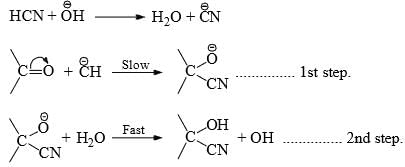
- Acid-catalysed-The acid protonates the negatively charged oxygen atom in the first step. The slow attack of the reagent to the carbonyl carbon in the second step completes the addition.
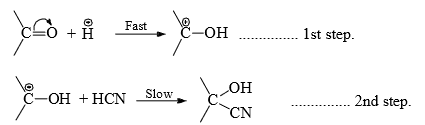
Although acids increase the catinoid character of the carbonyl carbon it reduces the concentration of the nucleophile, e.g.,
- Hence, a highly acidic medium retards the reaction. Weak acids which can from hydrogen bonds are used. In practice the carbonyl compound is mixed with sodium cyanide and acid is added to generate HCN. The acid should be insufficient to consume all the sodium cyanide so that the medium remains alkaline.
- In both acid and base-catalyzed additions, the nucleophile is added in the slowest step and hence the reaction is called nucleophile addition.
- The addition product of carbonyl compound with HCl is not isolable. The equilibrium lies far to the left since chlorine is a good leaving group.
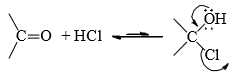
- A number of carbonyl addition reactions follow the same mechanistic pattern of cyanohydrin formation. A few of these are mentioned.
Alcohol Addition to Carbonyl Group
The addition of alcohol group to form hemiacetal is catalysed by both base and acid but the formation of acetal from hemiacetal is catalysed by acids only.
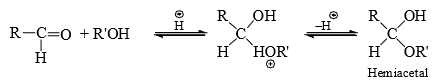
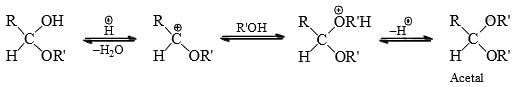
Bisulphite Addition to Carbonyl Group
Aldehydes, methyl ketones or cyclic ketones add with sodium bisulphite in aqueous medium to form crystalline adducts. The effective nucleophile is 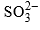 which is a strong nucleophile, and so no catalyst is required for the addition.
which is a strong nucleophile, and so no catalyst is required for the addition.
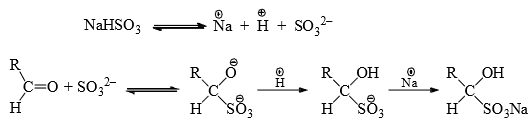
Carbanion Addition to Carbonyl Group
A large number of carbanion addition reactions are known. Addition of Grignard reagent is given for illustration. Grignard reagent may be considered as a carbanion donor due to the polarized carbon-metal bond 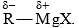
A straightforward reactions then possible, e.g.,
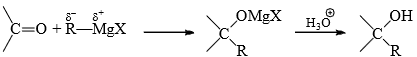
However, addition of MgBr2 (Lewis acid) increases the rate of addition. From this and other evidence (complex of Mg with the carbonyl oxygen), it is suggested that one molecule of RMgX complexes with the oxygen to facilitate the attack of the other molecule to the carbonyl carbon.
Substitution Reactions
A reaction in which one group or atom is replaced by another is called a substitution reaction. The incoming group is bonded to the same carbon to which the leaving group was bonded.
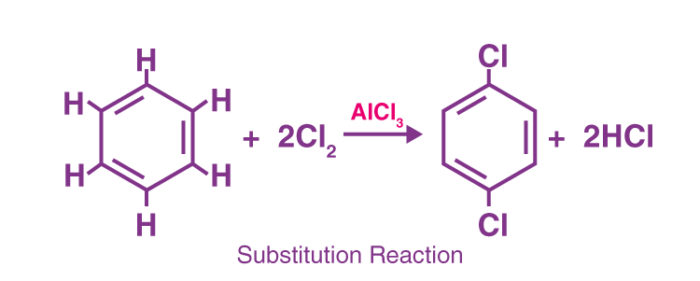
- Substitution reaction has been classified according to the nature of the substituents involved.
(i) Free radical substitution
(ii) Nucleophilic substitution
(iii) Electrophilic substitution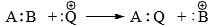
- It will be seen that in all types of substitutions the displaced species belong to the same class as the attacking species.
(i) Free Radical Substitution
Radical substitution reactions are initiated by radicals in the gas phase or in non-polar solvents.
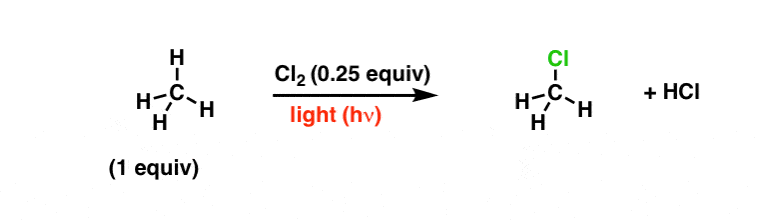
- Thus, methane and chlorine react in the presence of sunlight or heat to give methyl chloride.
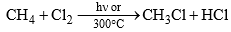
- Light energy or heart causes homolytic fission of chlorine producing chlorine radicals that attack methane to form methyl chloride.
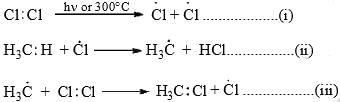
- The mechanism is supported by the fact that no reaction occurs in the dark, and in the presence of tetraethyl lead (0.02%) the reaction takes place at 140°C. Tetraethyl lead decomposes at 140°C to ethyl radical which produced chlorine radical from chlorine for the propagation of the reaction as given above.
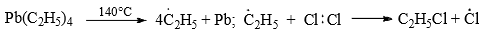
The reaction proceeds by the repetition of steps (ii) and (iii) - When the ratio of methane to chlorine is high, methyl chloride is formed predominantly and when chlorine is in excess all the hydrogens are replaced to give carbon tetrachloride
(ii) Nucleophilic Substitution
Nucleophilic substitution reaction is a class of organic reactions where one nucleophile replaces another.
- The group which takes electron pair and displaced from the carbon is known as the “leaving group” and the molecule on which substitution takes place known as “substrate”.
- The leaving group leaves as a neutral molecule or anion.
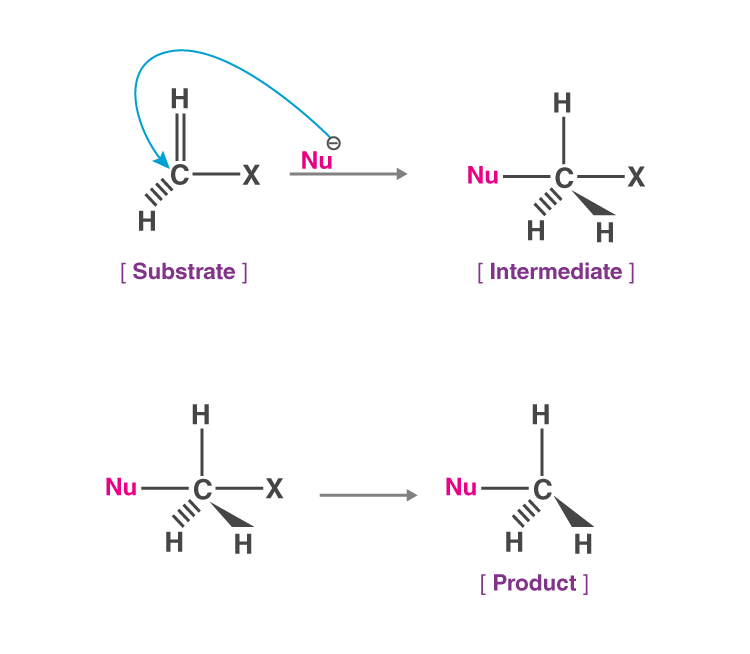
(a) At Saturated Carbon
- Nucleophilic substitution reaction involves the displacement of a nucleophile by another. These reactions have great synthetic importance. A classical example is the hydrolysis of alkyl halides.
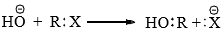
- The nucleophile furnishes as electron pair to the carbon from which the leaving group departs with the bonding pair of electrons. Investigations by ingold and co-workers indicate that nucleophile substitution reaction can proceed by two different paths which have been designated by ingold as SN1 and SN2 depending on the nature of the substrate, the nucleophile, the leaving group, and the solvent.
SN1 Mechanism
- Kinetic studies of the hydrolysis of t-butyl bromide indicate that the rate of the reaction is proportional to the concentration of the alkyl halide, i.e., 3 rate
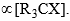
- Since the rate of reaction is dependent on one of the reactants, the reaction is a first-order reaction. Nucleophilic substitution reaction which follows first-order kinetics is designated SN1 (Substitution Nucleophilic Unimolecular).
- As the rate of reaction is independent of, it is interpreted that the halide undergoes slow ionization in the first step producing carbocation intermediate. In the second step, a rapid attack of
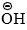 on the carbocation completes the hydrolysis.
on the carbocation completes the hydrolysis.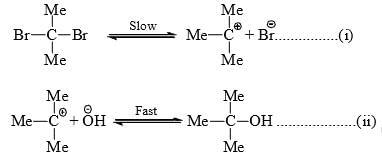
- The energy required for the ionization of the halide is supplied by the energy of solvation of the ions. Since a carbocation is formed in the slowest step, the alkyl halide which can most easily form a stable carbocation will favor hydrolysis by SN1 path. Hence, the order of hydrolysis of alkyl halides by SN1 path is:
Allyl, Benzyl > 3° > 2° > 1° > CH3X (ct. inductive effect and resonance effects) - Stereochemistry of SN1 reaction: Since a carbocation is flat (sp2, trigonal planar) with the vacant 2p orbital vertical to the plane bearing the three groups, the attack of the reagent can occur from either side of the plane with equal probability, i.e., the racemic product should result if the alkyl halide is chiral.
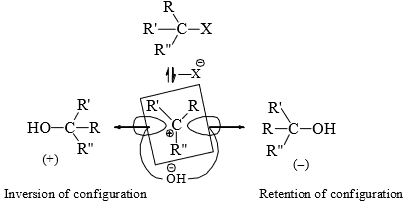
- Pure racemization (50/50 mixture) is rarely observed. This is because the leaving group lies close to the carbocation shielding the side from the attack till it has sufficiently moved away. Thus, more attacks of the reagent occurs from the opposite side to the leaving group. This causes more inversion than retention of configuration.
- Stable carbocations have a longer life to permit solvation from either side of the plane of the carbocation resulting in a greater proportion of racemization. Greater proportion of inversion is observed with more nucleophilic solvent due to faster attack from the opposite side to the leaving group.
SN2 Mechanism
- Nucleophilic substitution reactions which follow second-order kinetics is called SN2 (Substitution Nucleophilic Bimolecular) reaction. The rate of a second-order reaction depends upon the concentrations of both reactants.
- Thus, the rate of hydrolysis of methyl bromide with NaOH has been found to be of second-order, i.e.,
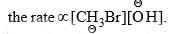
- Since the rate-determining step involves both CH3Br and, a collision between the two reactants resulting in the direct displacement of
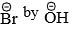 occurs in such a way that while a new C—OH bond is being formed, the reaction is a concerted one-step reaction without any intermediate.
occurs in such a way that while a new C—OH bond is being formed, the reaction is a concerted one-step reaction without any intermediate.
(b) At Unsaturated Carbon
- When a carbon atom multiply bonded to heteroatom (e.g., O, N, S, etc.), bears a good leaving group such as Cl, OH, OR, NH2, etc., nucleophilic attack on the carbon results in substitution rather than addition.
- Some examples are given for illustration.
(i) Hydrolysis of acid chloride, acid amide, and ester: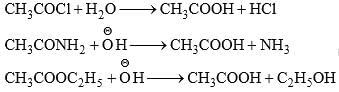
(ii) Formation of acid amide from acid chloride:
(iii) Ester from acid: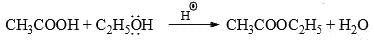
The general mechanism of the reactions is: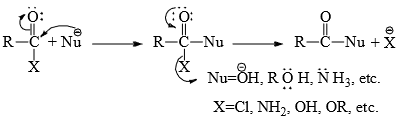
- The nucleophilic attack on the unsaturated carbon displaces an electron pair to the oxygen atom in the transition state. When the electron pair reverts, the leaving group is displaced.
(c) Automatic Nucleophilic Substitution
Substitution of Hydrogen of Benzene
- Nucleophilic substitution of the hydrogen of benzene is difficult because (a) the π-electron cloud will repel the nucleophile and (b) it would be difficult for the ring to accommodate the negative charge brought in by the nucleophile.
- However, the presence of a strong electron-withdrawing group may overcome the difficulties and nucleophilic substitution then is possible. Thus, nitrobenzene gives o-nitrophenol on treatment with strong NaOH in the presence of oxidizing agents such as KNO3 or K3Fe(CN)6.
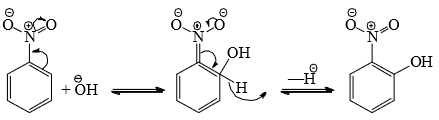
Since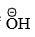 is a better leaving group than
is a better leaving group than 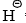 , the expulsion of
, the expulsion of 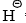 is aided by oxidation.
is aided by oxidation.
Substitution of Groups other than Hydrogen
- Nucleophilic substitution is observed in suitably substituted aromatic compounds. These reactions may be unimolecular (SN1) or bimolecular (SN2).
- SN1 type: A common example of aromatic nucleophilic substitution which resembles nucleophilic substitution at saturated carbon by SN1 path is the decomposition of diazonium salts in polar medium and formation of mixed products in the presence of various nucleophiles.
- The rate of reaction is found to be dependent on the concentration of ArN2– only, i.e., the reaction is unimolecular (SN1). Hence, it is presumed that in the first slow, rate-determining step, heterolysis of C—N bond takes place with the formation of nitrogen and aryl cation. This is followed by a fast attack of the nucleophile in the second step. The aryl cation is very reactive and takes up any nucleophile present, even recombines with the eliminated N2. Therefore, the reaction is reversible.
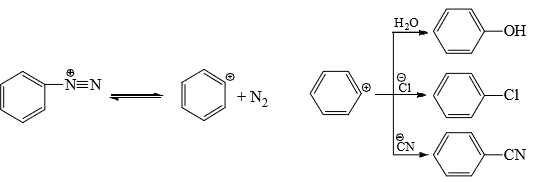
Although aryl cation is very unstable, the driving force for its formation is the elimination of very stable nitrogen (bond energy of N≡ N= 226 kcal/mol). - SN2 type: Halogens activated by strong electron-withdrawing groups from o-and p-positions undergo displacement by bimolecular mechanism.
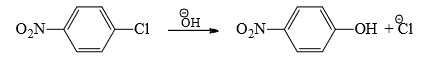
The rate of the reaction is proportional to the concentration of both the reactants, i.e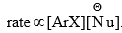 The reaction fails in the absence of the electron-withdrawing group, NO2. Hence, activation of the halogen by the NO2 group is suggested.
The reaction fails in the absence of the electron-withdrawing group, NO2. Hence, activation of the halogen by the NO2 group is suggested. - The carbon-bearing halogen is electron-deficient due to the electron withdrawal by the NO2 and Cl groups. This promotes attack by the nucleophile. The intermediate formed on the attack of the nucleophile is stabilized by delocalization of the negative charge by the NO2 group. The intermediate then passes to a more stable product by the expulsion of the halogen.
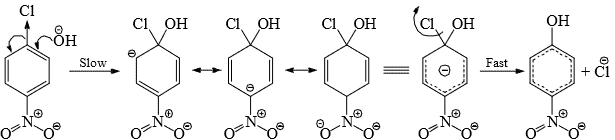
- The reaction is not exactly analogous to aliphatic SN2 reaction in the following:
(i) The attack of the nucleophilic occurs from the side as the back-side attack is sterically hindered by the ring.
(ii) The carbon bearing the leaving group forms four rather than five bonds as in a normal SN2 reaction.
(iii) The bond to the attacking nucleophile is first formed and then the bond to the leaving group breaks. i.e., it an addition-elimination process.
(iv) An intermediate rather than a transition state is formed. Hence, the reaction is designated SN2 (aromatic).
Substitution of unactivated halogens (Benzyne mechanism)
- Unactivated halogens in aromatic compounds undergo indirect nucleophilic displacement in the presence of very strong bases such as sodamide in liquid ammonia at low temperature (–33°C) or strong NaOH at high temperature (340°C).
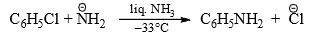
- It has been observed that
 is able to remove a proton from the benzene ring. It is, therefore, suggested that
is able to remove a proton from the benzene ring. It is, therefore, suggested that  removes a proton from the ortho positive to chlorine. This results in the simultaneous expulsion of Cl and formation of benzyne* intermediate. Benzyne is exceedingly reactive and reacts with any nucleophile present, in this case, the solvent liquid ammonia, to form an additional product.
removes a proton from the ortho positive to chlorine. This results in the simultaneous expulsion of Cl and formation of benzyne* intermediate. Benzyne is exceedingly reactive and reacts with any nucleophile present, in this case, the solvent liquid ammonia, to form an additional product.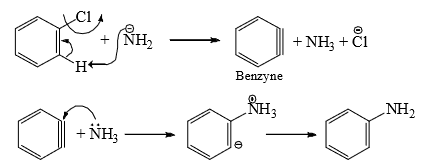
- The reaction is, therefore, an elimination-addition process. The symmetrical benzyne intermediate can be attacked at either of the carbons forming the triple bond. This is observed in chlorobenzene with labeled carbon (14C) bearing the chlorine.
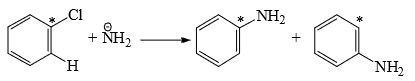
- Thus, p-chlorotoluene on treatment with strong NaOH at 340°C gives a mixture of o-and p-cresols. This cine substitution (aromatic nucleophilic substitution at a different position from the leaving group) supports the formation of benzyne intermediate. Further support of the mechanism comes from the observation that no reaction takes place when both the ortho positions are substituted.
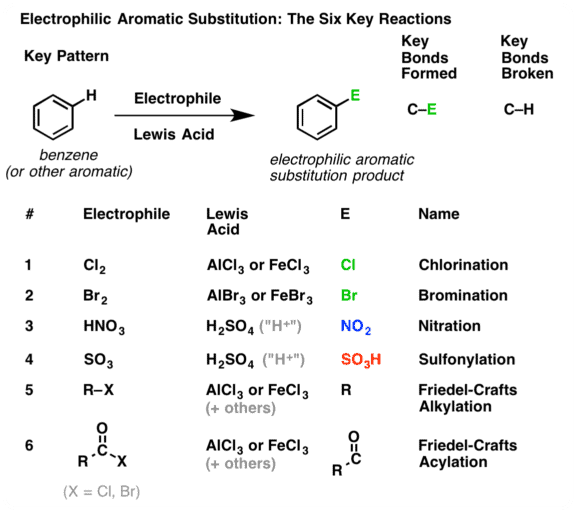
Electrophilic (Aromatic) Substitution
Electrophilic aromatic substitution reactions are organic reactions wherein an electrophile replaces an atom which is attached to an aromatic ring. Commonly, these reactions involve the replacement of a hydrogen atom belonging to a benzene ring with an electrophile.
- We know that benzene is a resonance hybrid which is a flat ring with a cyclic cloud of a negative charge, above and below the plane of the ring.
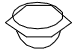
- The benzene ring, therefore, discourages nucleophilic attack and encourages electrophilic attack. Alkenes also encourage electrophilic attack resulting in addition while the electrophilic attack on benzene results in substitution since addition would involve loss of resonance stabilization of benzene.
- Acetylenic bound in benzyne is unlikely since the ring will be greatly deformed with accompanying strain. It has been suggested that the third bound is formed by the overlap of the two sp2 orbitals vacated by hydrogen and chlorine. The overlap of the two sp2 orbitals will be poor and consequently, benzyne will be very reactive.
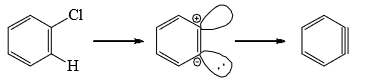
Its existence has been established by trapping experiments and spectroscopy.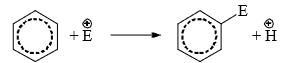
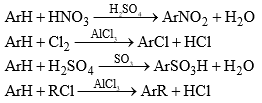
- Hence, the typical reactions of benzene and its derivations are electrophilic substitution. Aromatic electrophilic substitution includes a wide variety of reactions, e.g., nitration, sulphonation, halogenation, Friedel-Crafts reactions, etc. The reaction permits the direct introduction of groups to the ring and their subsequent transformation to various other products.
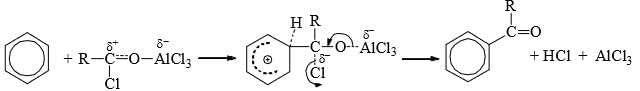
Mechanism
The various aromatic electrophilic substitutions follow the same mechanistic pattern which is generalized below.
- The reagent and the catalyst (Lewis acids or proton acids) undergo an acid-base reaction to produce the attacking electrophile which then attacks the ring to form a carbocation (σ complex) in the first slow step. In the second step, the proton from the site of the attack is rapidly removed by the base to complete the reaction.
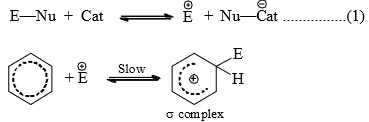
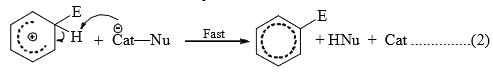
- The energy gained in passing from the complex to the product (regain of aromaticity) provides the energy for breaking the strong C—H bond. It may seem that the formation of the σ complex would involve high energy. This is, however, not so because the energy liberated by the formation of a C—E bond and the delocalization of the positive charge lowers the energy of the σcomplex.
- The rate of reaction does not involve the breaking of C—H bond. This has been established by the isotope effect. The rates of reactions are the same on the replacement of hydrogen by deuterium or tritium. Hence, the rate of the substitution reaction is determined by the slow attachment of the electrophilic to the ring in the first step.
Let us now consider some of the individual substitution reactions.
Nitration
- Nitration of benzene is effected by a mixture of concentrated nitric and sulphuric acids. In the absence of sulphuric acid the reaction in slow. It is suggested that H2SO4 acts as strong acid and protonates HNO3 to generate nitronium ion,
 . This has been confirmed by various methods.
. This has been confirmed by various methods.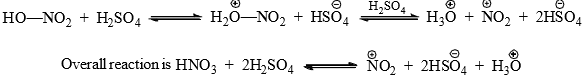
- The nitronium ion then attacks the benzene ring to form a carbocation in the first step.
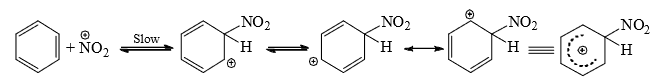
- In the second step, a fast abstraction of hydrogen from the site of attack by the base
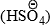 completes the reaction.
completes the reaction. - The presence of appreciable amount of water in the acid mixture is not desirable since it reduces the nitronium ion; hence, the use of the concentrated acid mixture.
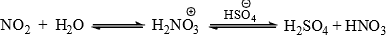
- Phenol, which is highly reactive due to the mesomeric effect of the OH group. is nitrated even with dilute HNO3. A small amount of phenol is oxidized to produce HNO2 which with HNO3 give nitrosonium ion,
 . The latter nitrostates the reactive phenol to yield nitrophenol which is rapidly oxidized to nitrophenol with HNO3 and nitrous acid is produced by reduction of HNO3. With the progress of the reaction more and more nitrous acid is produced and the reaction rate is increased.
. The latter nitrostates the reactive phenol to yield nitrophenol which is rapidly oxidized to nitrophenol with HNO3 and nitrous acid is produced by reduction of HNO3. With the progress of the reaction more and more nitrous acid is produced and the reaction rate is increased.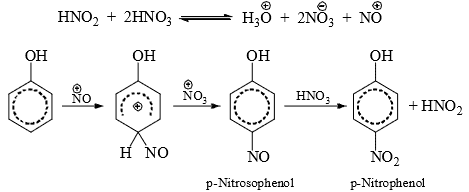
Sulphonation
- Benzene on heating with concentrated or better with fuming sulphuric acid gives benzene sulphonic acid, C6H5SO3H. Sulphonation can also be affected with ClSO3H.
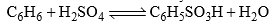
- The reaction is slow with concentrated H2SO4 but rapid with oleum or SO3 in inert solvent. The electrophile is SO3 which is present in a small amount in H2SO4.
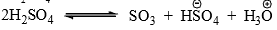
- The electrophile, SO3, is neutral but has a powerful electrophilic sulfur which attacks the ring.
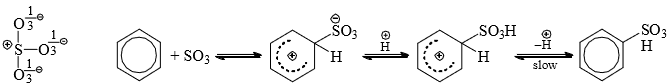
- The rate-determining step involves the breaking of C—H bond since deuterated benzene is sulfonated more slowly (isotopic effect). The reaction is reversible. On treatment with steam. SO3H group is replaced by hydrogen. The reversibility of the reaction has practical utility.
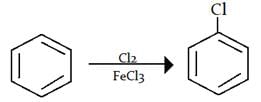
Halogenation
- Free halogens can attack activated benzene rings but Lewis acid catalyst is required for benzene rings.
- It is suggested that probably benzene first forms a π complex with the halogen molecule, e.g., Br2. The Lewis acid (FeBr3) then polarizes the Br—Br bond and helps in the formation of a σcomplex between benzene carbon and the electrophilic end of the polarized bromine by removing the incipient bromide ion. Subsequent abstraction of hydrogen in the second step completes the reaction.
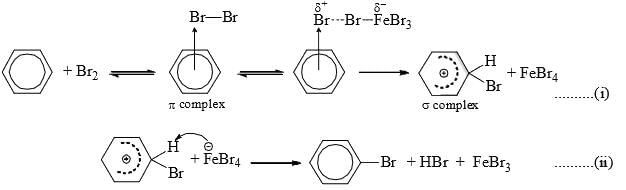
- The order of reactivity of the halogens is F2 > Cl2 > Br2 > I2. Fluorine is too reactive for practical use. Under ordinary conditions, iodination fails. In the presence of HNO3 direct iodination has been affected. The attacking electrophile
 is produced by HNO3.
is produced by HNO3.
Friedel-Crafts Reaction
- Alkylation and acylation of aromatic rings with alkyl halides and acid chlorides or anhydrides respectively in the presence of Lewis acids, e.g., AlCl3, FeBr3, BF3, BCl3, etc. is known as Friedel-Crafts reaction.
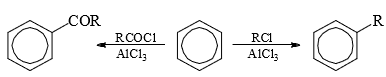
- Alkylation: The function of the catalyst is to provide a real or potential carbocation for the electrophilic attack. When the alkyl group can form a stable carbocation as in the case of 3° halides, the attacking species is the real carbocation which forms an ion pair (23).
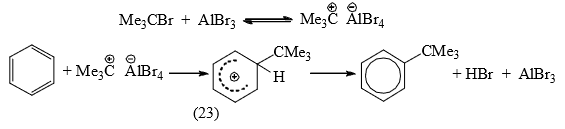
In other cases, a polarized completed with a potential carbocation is the attacking species.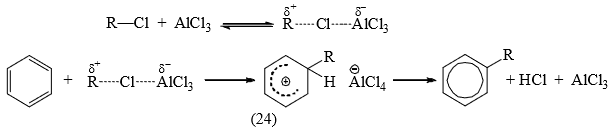
The intermediates (23) and (24) have been isolated in some cases. It has been observed that the same alkyl halide gives a rearrangement and an unrearranged product with a different Lewis acid catalyst.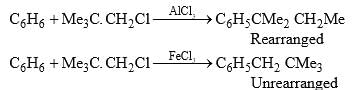
Since AlCl3 is a stronger Lewis acid than FeCl3, it is presumed that the complex with AlCl3 is so strongly polarized that the alkyl group attains almost a free carbocation character to undergo rearrangement.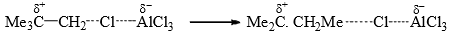
The alkylating reagent besides alkyl halides may be aliphatic alcohols, alkenes, ethers, etc., in the presence of strong proton acids which generate the carbocation for the electrophilic attack. - Acylation: Acylating reagents are acid chlorides or acid anhydrides in the presence of Lewis acid. The electrophilic species may be (a) acyl cation as an ion pair or (b) a polarized 1:1 complex. Probably both mechanisms operate depending on conditions.
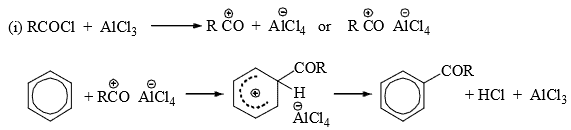
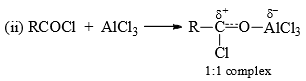
- In both cases, one mole of the catalyst remains complexed with the product, ketone. Hence, slightly more than one mole of Lewis acid is required for acylation. Acylation may be affected with acid anhydrides also. The attacking species in this case may be free
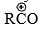 or RCOCl. (For further details see Friedel-Crafts reaction.
or RCOCl. (For further details see Friedel-Crafts reaction.
|
39 videos|92 docs|46 tests
|
FAQs on Reaction Mechanism Chemistry - Organic Chemistry
| 1. What is the difference between addition reactions and substitution reactions in chemistry? |  |
| 2. How do addition reactions occur in organic chemistry? | |
| 3. What is the mechanism behind substitution reactions in chemistry? | |
| 4. Can you provide an example of an addition reaction in organic chemistry? | |
| 5. What are some common reagents used in substitution reactions? | |




Semester Notes
,Free
,Extra Questions
,Reaction Mechanism Chemistry | Organic Chemistry
,Viva Questions
,past year papers
,Reaction Mechanism Chemistry | Organic Chemistry
,Exam
,mock tests for examination
,ppt
,Previous Year Questions with Solutions
,shortcuts and tricks
,practice quizzes
,Summary
,video lectures
,MCQs
,Important questions
,study material
,Reaction Mechanism Chemistry | Organic Chemistry
,Objective type Questions
,Sample Paper
;


Reaction Mechanism Chemistry Free PDF Download
Importance of Reaction Mechanism Chemistry
Reaction Mechanism Chemistry Notes
Reaction Mechanism Chemistry Chemistry Questions
Study Reaction Mechanism Chemistry on the App
|
© EduRev
|
Education Revolution
|



|





