Organic Reactions With Mechanism (Part - 4) | Organic Chemistry PDF Download

Schmidt Reaction
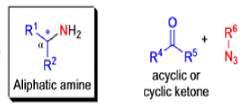
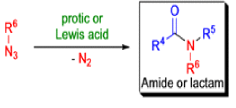
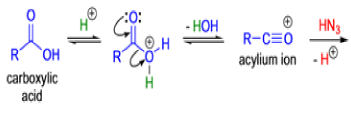
The Schmidt reaction is an organic reaction in which an azide reacts with a carbonyl derivative, usually a aldehyde, ketone, or carboxylic acid, under acidic conditions to give an amine or amide, with expulsion of nitrogen.
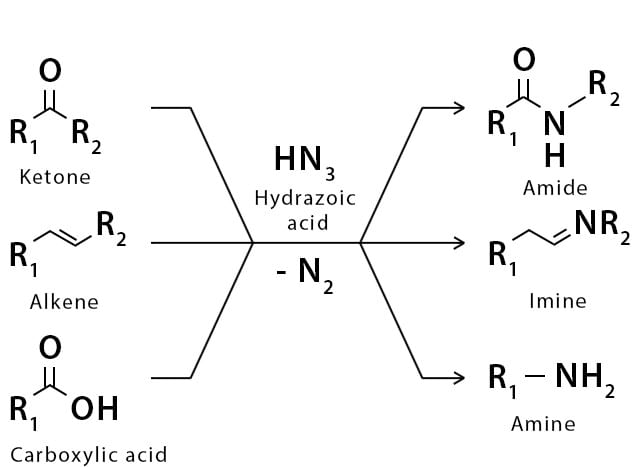
- In 1923, Schmidt reported that heating hydrazoic acid (HN3) with benzophenone in the presence of sulfuric acid, afforded benzanilide in quantitative yield.
- Later this transformation was shown to be general for ketones, aldehydes, and carboxylic acids that underwent similar reactions with HN3 to give amides, nitriles, and amines, respectively.
- The reaction of carbonyl compounds with hydrazoic acid or alkyl azides in the presence of acid catalysts is known as the Schmidt reaction.
- The general features of the Schmidt reaction are:
- Protic acids are used as acid catalysts (e.g., H2SO4, PPA, trichloroacetic acid/H2SO4, TFA, TFAA), and sulfuric acid is by far the most widely used;
- hydrogen azide is handled either as a solution in an inert solvent (e.g., CHCl3) or generated in situ by adding NaN3 to the acidic reaction mixture;
- HN3 is known to be toxic and explosive (especially on large scale).
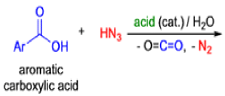

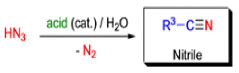
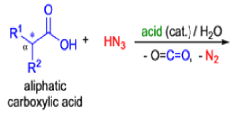

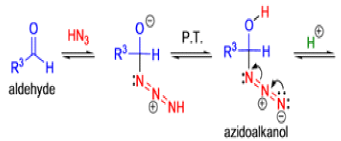
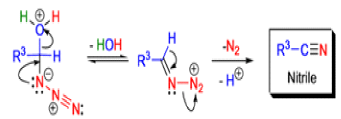
Mechanism
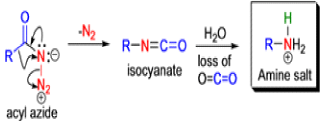
Darzens Glycidic Ester Condensation
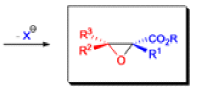
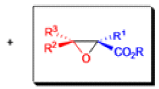
The formation of α, β epoxy esters (glycidic esters) from aldehydes and ketones and α halo esters under basic conditions is known as the Darzens glycidic ester condensation.
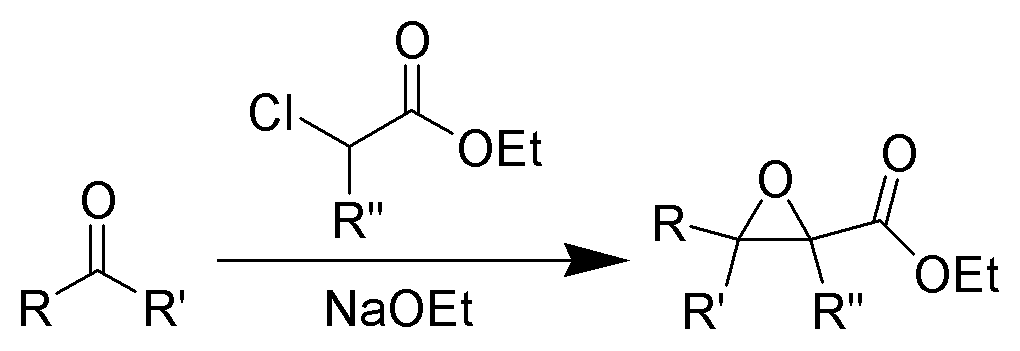
- The reaction is general, since aromatic aldehydes and ketones, aliphatic ketones as well as α,βunsaturated and cyclic ketones react smoothly and give good yields of the expected glycidic esters.
- Aliphatic aldehydes usually give lower yields, but the deprotonation of the αhalo ester with a strong kinetic base prior to the addition of the aldehyde results in acceptable yields. α-Chloro esters are preferable to Bromo or iodo esters, since they give higher yields.
- In addition to αhalo esters, αhalo sulfones, nitriles, ketones, and amides can also be used to obtain the corresponding glycidic derivatives.
- Glycidic esters are versatile synthetic intermediates: the epoxide functionality can be opened with various nucleophiles and upon thermolysis, the intermediates undergo decarboxylation to afford the corresponding one carbon homologue of the starting aldehyde or ketone.

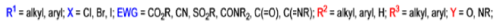

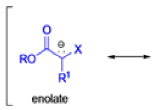
Mechanism

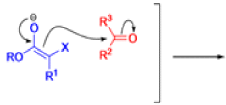
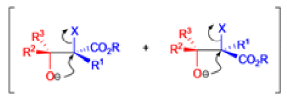
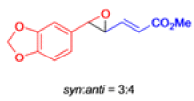
Claisen Rearrangement
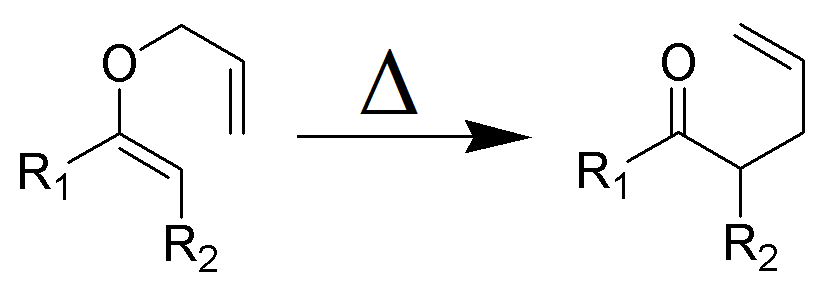
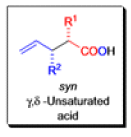
The Claisen rearrangement is a powerful carbon –carbon bond -forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl.
- In 1912, L. Claisen described the rearrangement of allyl phenyl ethers to the corresponding C-allyl phenols and also described the transformation of O-allylated acetoacetic ester to its C-allylated isomer in the presence of ammonium chloride upon distillation.
- Named after its discoverer, the thermal [3, 3]-sigmatropic rearrangement of allyl vinyl ethers to the corresponding γ,δunsaturated carbonyl compounds is called the Claisen rearrangement.
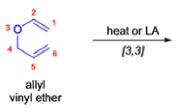
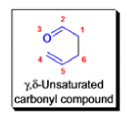
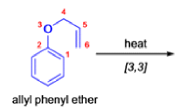
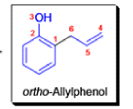
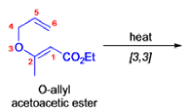
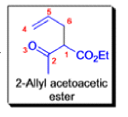
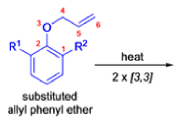
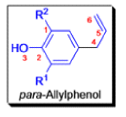
The Claisen, Johnson–Claisen, Ireland–Claisen, para-Claisen rearrangements, along with the Carroll rearrangement belong to the category of [3,3]-sigmatropic rearrangements. The Claisen rearrangement is a concerted process and the arrow pushing here is merely illustrative.
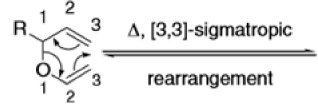
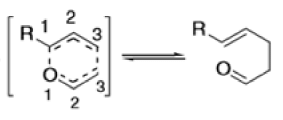
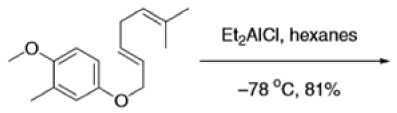
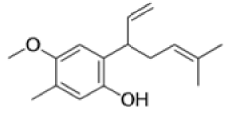
Mechanism
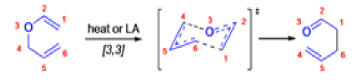
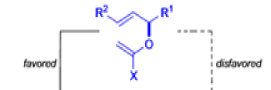
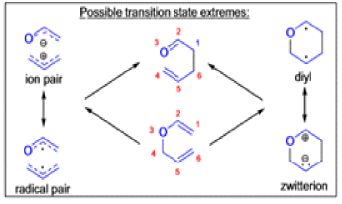
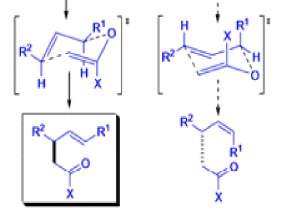
Claisen-Ireland Rearrangement
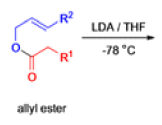
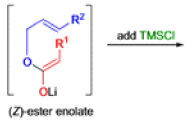
The IrelandClaisen rearrangement is an organic reaction used to convert an allyl ester to a γ,δ-unsaturated carboxylic acid using LDA, TMSCl, and NaOH/H2O.
- The reaction begins with deprotonation of the α-hydrogen of the easter to form an enolate which then attacks TMSCl to stabilize the charge.
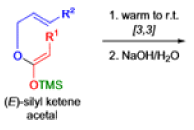
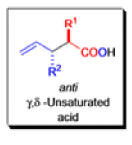
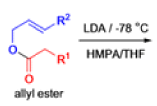
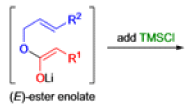
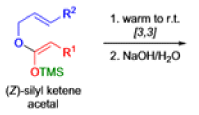
Cope Rearrangement
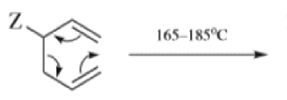
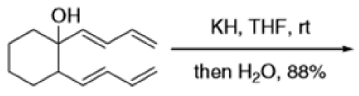
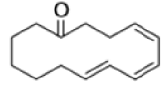
When 1,5-dienes are heated, the Cope, oxy-Cope, and anionic oxy-Cope rearrangements belong to the category of [3,3]-sigmatropic rearrangements occurs to generate an isomeric 1,5-diene.
- Since it is a concerted process, the arrow pushing here is only illustrative.
- When the diene is symmetrical about the 3,4 bond, we have the unusual situation where a reaction gives aproduct identical with the starting material:

- Therefore, a Cope rearrangement can be detected only when the diene is not symmetrical about this bond.
- Any 1,5-diene gives the rearrangement; for example, 3-methyl-1,5-hexadiene heated to 300 0C gives 1,5-heptadiene.
- However, the reaction takes place more easily (lower temperature required) when there is a group on the 3- or 4-carbon with leads to the new double bond being substituted.
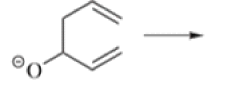
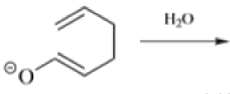
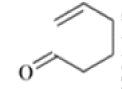
- The reaction is obviously reversible and produces an equilibrium mixture of the two 1,5-dienes, which is richer in the thermodynamically more stable isomer. However, the equilibrium can be shifted to the right for 3-hydroxy-1,5-dienes, because the product tautomerizes to the ketone or aldehyde:
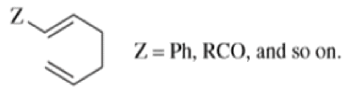

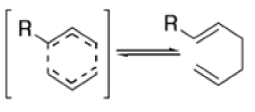
Oxy-cope rearrangement:
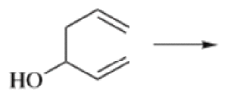

The reaction of 3-hydroxy-1,5-dienes is called the oxy-Cope rearrangement,and has proved highly useful in synthesis.
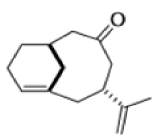
The oxy-Cope rearrangement is greatly accelerated, if the alkoxide is used rather than the alcohol, where the direct product is the enolate ion, which is hydrolyzed to the ketone.
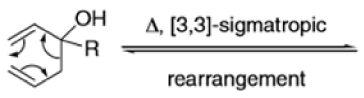
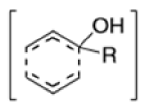
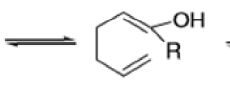

Anionic oxy-cope rearrangement:
Chichibabin Amination Reaction
The Chichibabin reaction is a method for producing 2-aminopyridine derivatives by the reaction of pyridine with sodium amide.
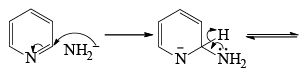
- The direct amination of pyridine with sodium amide takes place in liquid ammonia.
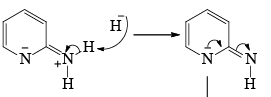
- Following the addition elimination mechanism first a nucleophilic NH2– is added while a hydride (H–) is leaving.
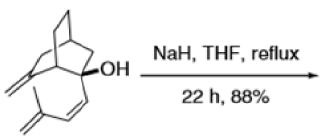
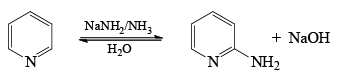
Mechanism
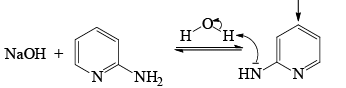
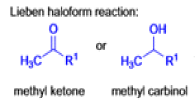
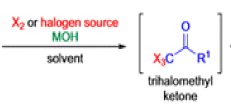
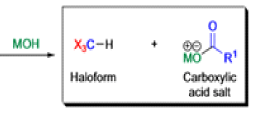
Haloform Reaction
The haloform reaction is a chemical reaction where a haloform (CHX3, where X is a halogen) is produced by the exhaustive halogenation of a methyl ketone (RCOCH3, where R can be either a hydrogen atom, an alkyl or an aryl group), in the presence of a base.
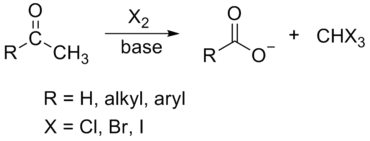
The general features of this reaction are:
- compounds containing the methyl ketone (CH3-CO) functional group or compounds that get oxidized under the reaction conditions to methyl ketones will undergo the transformation;
- in addition to methyl ketones and methyl carbinols, mono-, di-, and trihalogenated methyl ketones also give rise to haloforms;
- the reaction is usually conducted in aqueous alkali, but for compounds that are insoluble in water the addition of a co-solvent such as dioxane or THF is necessary;
- the halogen can be chlorine, bromine, and iodine, but elemental fluorine gas cannot be used due to its immense reactivity;
- the reaction is sensitive to steric hindrance, so when the R1 group is bulky, the hydrolysis of the trihalomethyl ketone usually does not take place, and the reaction stops; certain side reactions such as the αhalogenation and subsequent cleavage of the other alkyl group is possible.
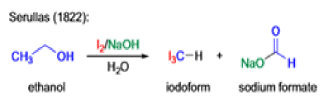
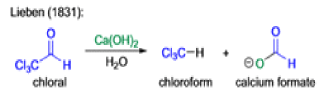

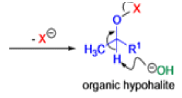
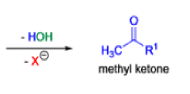
Mechanism


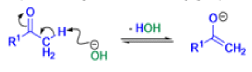
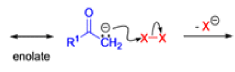
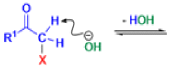
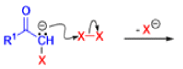
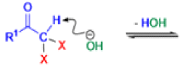
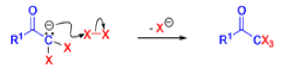

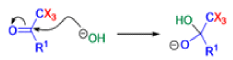
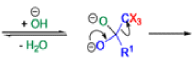
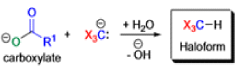
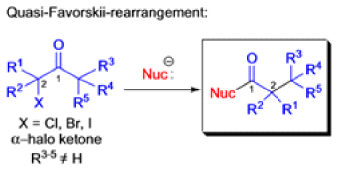
Favorskii And Homo-favorskii Rearrangement
The Favorskii rearrangement is principally a rearrangement of cyclopropanones and α-halo ketones that leads to carboxylic acid derivatives.
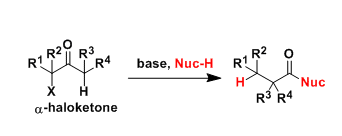
- Treatment of αhalo ketones possessing at least one αhydrogen with base in the presence of a nucleophile (alcohol, amine, or water) results in a skeletal rearrangement via a cyclopropanone intermediate to give carboxylic acids or carboxylic acid derivatives (esters or amides).
- This reaction is known as the Favorskii rearrangement, and it is widely used for the synthesis of highly branched carboxylic acids. The halogen substituent can be a chlorine, bromine or iodine, while the base is usually an alkoxide or hydroxide.
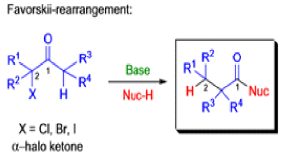
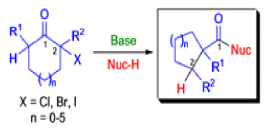
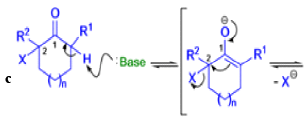
Mechanism
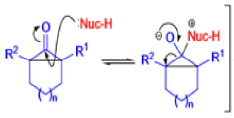
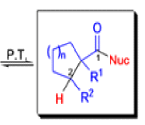
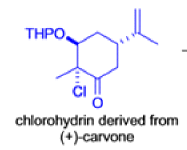

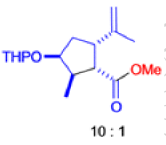
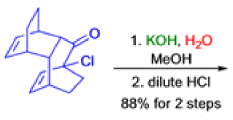
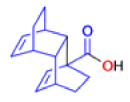
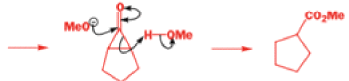
The Favorskii Rearrangement
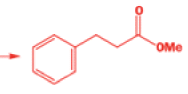
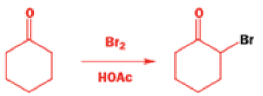
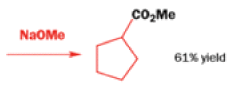
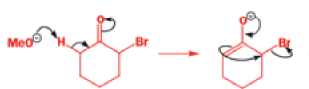
We hope you have appreciated the smooth mechanistic progression so far in this chapter, from Wagner– Meerwein to pinacol and semipinacol through dienone–phenol to benzilic acid. Our aim is to help you gain an overall view of the types of rearrangements that take place (and why) and not to present you with lots of disconnected facts. It is at this point, however, that our mechanistic journey takes a hairpin bend. A surprising one, too, because, when we show you the Favorskii rearrangement, you would be forgiven for wondering what the fuss is about: surely it’s rather like a variant of the benzilic acid rearrangement?
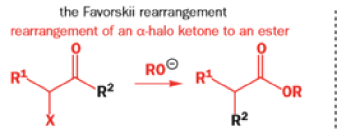
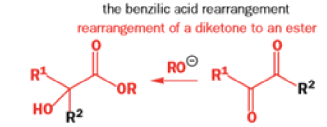


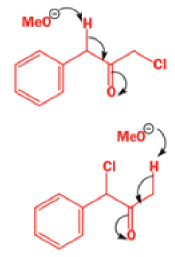
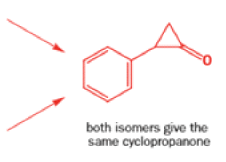
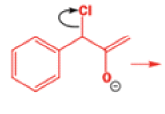
Well, this is what chemists thought until 1944, when some Americans found that two isomeric αchloro ketones gave exactly the same product on treatment with methoxide. They suggested that both reactions went through the same intermediate.
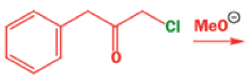
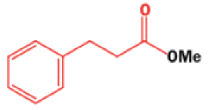
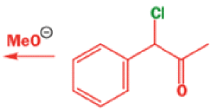
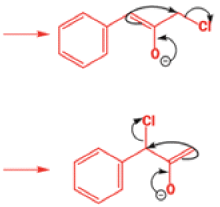
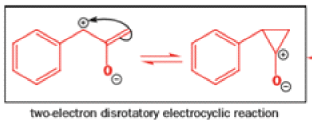
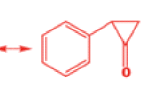
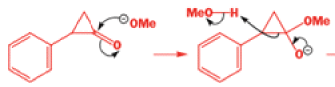
That intermediate is a three-membered cyclic ketone, a cyclopropanone: the alkoxide acts not as a nucleophile (its role in the benzilic acid rearrangement) but as a base, enolizing the ketone. The enolate can alkylate itself intramolecularly in a reaction that looks bizarre but that many chemists think is not unreasonable. The product is the same cyclopropanone in each case.
|
44 videos|102 docs|52 tests
|
FAQs on Organic Reactions With Mechanism (Part - 4) - Organic Chemistry
| 1. What is the Schmidt reaction? |  |
| 2. How does the Darzens glycidic ester condensation work? | |
| 3. What is the Claisen rearrangement? | |
| 4. How does the Cope rearrangement work? | |
| 5. What is the haloform reaction? | |




Organic Reactions With Mechanism (Part - 4) | Organic Chemistry
,Viva Questions
,video lectures
,Semester Notes
,Summary
,shortcuts and tricks
,Important questions
,Exam
,Sample Paper
,ppt
,past year papers
,Organic Reactions With Mechanism (Part - 4) | Organic Chemistry
,study material
,Extra Questions
,practice quizzes
,MCQs
,Previous Year Questions with Solutions
,Objective type Questions
,mock tests for examination
,Organic Reactions With Mechanism (Part - 4) | Organic Chemistry
,Free
;






Organic Reactions With Mechanism (Part - 4) Free PDF Download
Importance of Organic Reactions With Mechanism (Part - 4)
Organic Reactions With Mechanism (Part - 4) Notes
Organic Reactions With Mechanism (Part - 4) Chemistry Questions
Study Organic Reactions With Mechanism (Part - 4) on the App
|
© EduRev
|
Education Revolution
|



|





