Organic Reactions With Mechanism and Applications (Part -2) | Organic Chemistry PDF Download

⇒Hofmann rearrangement:
The Hofmann rearrangement is the organic reaction of a primary amide to a primary amine with one fewer carbon atom.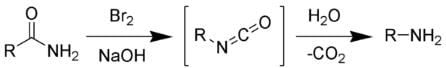 The Hofmann rearrangement.
The Hofmann rearrangement.
Mechanism:
The reaction of bromine with sodium hydroxide forms sodium hypobromite in situ, which transforms the primary amide into an intermediate isocyanate. The formation of an intermediate nitrene is not possible because it implies also the formation of a hydroxamic acid as a byproduct, which has never been observed. The intermediate isocyanate is hydrolyzed to a primary amine, giving off carbon dioxide.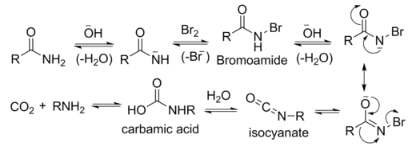
- Base abstracts an acidic N-H proton, yielding an anion.
- The anion reacts with bromine in an α-substitution reaction to give an N-bromoamide.
- Base abstraction of the remaining amide proton gives a bromoamide anion.
- The bromoamide anion rearranges as the R group attached to the carbonyl carbon migrates to nitrogen at the same time the bromide ion leaves, giving an isocyanate.
- The isocyanate adds water in a nucleophilic addition step to yield a carbamic acid (aka urethane).
- The carbamic acid spontaneously loses CO2, yielding the amine product.
Variations:
Several reagents can be substituted for bromine. Sodium hypochlorite, lead tetraacetate, N-bromosuccinimide, (bis(trifluoroacetoxy)iodo)benzene, and 1, 8-diazabicyclo [5.4.0] undec-7-ene (DBU) can effect a Hofmann rearrangement. In the following example, the intermediate isocyanate is trapped by methanol, forming a carbamate.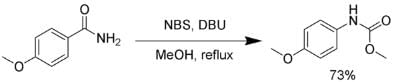 The Hofmann rearrangement using NBS.
The Hofmann rearrangement using NBS.
In a similar fashion, the intermediate isocyanate can be trapped by tert-butyl alcohol, yielding the tert-butoxycarbonyl (Boc)-protected amine.
The Hofmann Rearrangement also can be used to yield carbamates from α,β-unsaturated or α-hydroxy amides or nitriles from α,β-acetylenic amides in good yields (≈70%).
For amiloride, hypobromous acid was used to effect a Hofmann rearrangement.
Applications
- Aliphatic & aromatic amides are converted into aliphatic and aromatic amines, respectively
- In the preparations of anthranilic acid from phthalimide
- Nicotinic acid is converted into 3-Aminopyridine
- The symmetrical structure of α-phenyl propanamide does not change after Hofmann reaction.
- Gabapentin from mono-amidation 1, 1-cyclohexane diacetic acid anhydride with ammonia to 1, 1-cyclohexane diacetic acid mono-amide; followed by ‘Hoffmann’ rearrangement: U.S. Patent 20,080,103,334
⇒Curtius rearrangement:
The Curtius rearrangement (or Curtius reaction or Curtius degradation), first defined by Theodor Curtius in 1885, is the thermal decomposition of an acyl azide to an isocyanate with loss of nitrogen gas. The isocyanate then undergoes attack by a variety of nucleophiles such as water, alcohols and amines, to yield a primary amine, carbamate or urea derivative respectively. Several reviews have been published.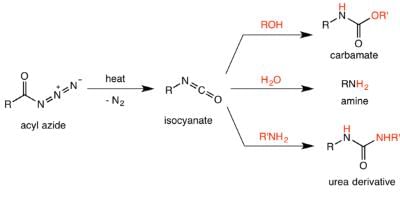 Preparation of acyl azide:
Preparation of acyl azide:
The acyl azide is usually made from the reaction of acid chlorides or anydrides with sodium azide or trimethylsilyl azide. Acyl azides are also obtained from treating acylhydrazines with nitrous acid. Alternatively, the acyl azide can be formed by the direct reaction of a carboxylic acid with diphenylphosphoryl azide (DPPA).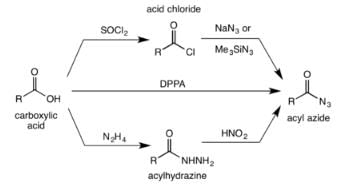
Reaction mechanism:
It was believed that the Curtius rearrangement was a two-step processes , with the loss of nitrogen gas forming an acyl nitrene, followed by migration of the R-group to give the isocyanate. However, recent research has indicated that the thermal decomposition is a concerted process, with both steps happening together, due to the absence of any nitrene insertion or addition byproducts observed or isolated in the reaction. Thermodynamic calculations also support a concerted mechanism.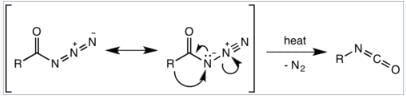 Mechanism of the Curtius rearrangement The migration occurs with full retention of configuration at the R-group. The migratory aptitude of the R-group is roughly tertiary > secondary ~ aryl > primary. The isocyanate formed can then be hydrolyzed to give a primary amine, or undergo nucleophilic attack with alcohols and amines to form carbamates and urea derivatives respectively.
Mechanism of the Curtius rearrangement The migration occurs with full retention of configuration at the R-group. The migratory aptitude of the R-group is roughly tertiary > secondary ~ aryl > primary. The isocyanate formed can then be hydrolyzed to give a primary amine, or undergo nucleophilic attack with alcohols and amines to form carbamates and urea derivatives respectively.
Synthetic applications:
The Curtius rearrangement is tolerant of a large variety of functional groups, and has significant synthetic utility, as many different groups can be incorporated depending on the choice of nucleophile used to attack the isocyanate.
For example, when carried out in the presence of tert-butanol, the reaction generates Boc-protected amines, useful intermediates in organic synthesis. Likewise, when the Curtius reaction is performed in the presence of benzyl alcohol, Cbz-protected amines are formed.
The Curtius rearrangement is used in the syntheses of the drugs tranylcypromine, candesartan, bromadol, terguride, benzydamine, gabapentin, igmesine and tecadenoson.
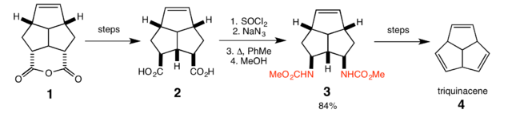
Triquinacene
R. B. Woodward et al. used the Curtius rearrangement as one of the steps in the total synthesis of the polyquinane triquinacene in 1964. Following hydrolysis of the ester in the intermediate (1), a Curtius rearrangement was effected to convert the carboxylic acid groups in (2) to the methyl carbamate groups (3) with 84% yield. Further steps then gave triquinacene (4).
Oseltamivir
In their synthesis of the antiviral drug oseltamivir, also known as Tamiflu, Ishikawa et al. used the Curtius rearrangement in one of the key steps in converting the acyl azide to the amide group in the target molecule. In this case, the isocyanate formed by the rearrangement is attacked by a carboxylic acid to form the amide. Subsequent reactions could all be carried out in the same reaction vessel to give the final product with 57% overall yield. An important benefit of the Curtius reaction highlighted by the authors was that it could be carried out at room temperature, minimizing the hazard from heating. The scheme overall was highly efficient, requiring only three “one-pot” operations to produce this important and valuable drug used for the treatment of avian influenza.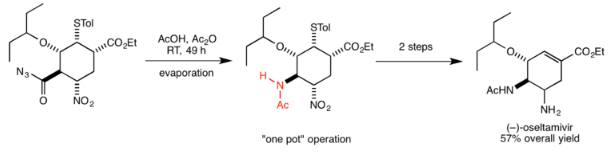
Dievodiamine
Dievodiamine is a natural product from the plant Evodia rutaecarpa, which is widely used in traditional Chinese medicine. Unsworth et al.’s protecting group-free total synthesis of dievodiamine utilizes the Curtius rearrangement in the first step of the synthesis, catalyzed by boron trifluoride. The activated isocyanate then quickly reacts with the indole ring in an electrophilic aromatic substitution reaction to give the amide in 94% yield, and subsequent steps give dievodamine.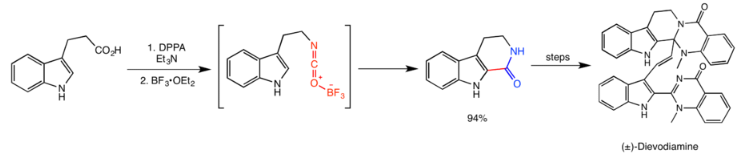
⇒ Lossen rearrangement:
The Lossen rearrangement is the conversion of a hydroxamic acid (1) to an isocyanate (3) via the formation of an O-acyl, sulfonyl, or phosphoryl intermediate hydroxamic acid O-derivative (2) and then conversion to its conjugate base. Here, 4-toluenesulfonyl chloride is used to form a sulfonyl O-derivative of hydroxamic acid.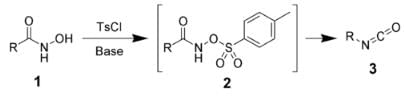 The isocyanate can be used further to generate ureas in the presence of amines (4) or generate amines in the presence of H2O (5).
The isocyanate can be used further to generate ureas in the presence of amines (4) or generate amines in the presence of H2O (5).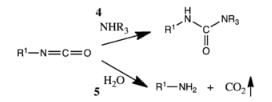
Reaction mechanism:
The mechanism below begins with an O-acylated hydroxamic acid derivative that is treated with base to form an isocyanate that generates an amine and CO2 gas in the presence of H2O. The hydroxamic acid derivative is first converted to its conjugate base by abstraction of a hydrogen by a base. Spontaneous rearrangement kicks off a carboxylate anion to produce the isocyanate intermediate. The isocyanate in the presence H2O hydrolyzes and then decarboxylation via abstraction of a hydrogen by a base generates an amine and CO2 gas.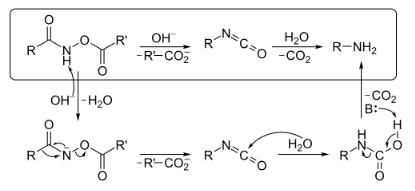 Hydroxamic acids are commonly synthesized from their corresponding esters.
Hydroxamic acids are commonly synthesized from their corresponding esters.
⇒ Simmons–Smith reaction:
The Simmons–Smith reaction is an organic cheletropic reaction involving an organozinc carbenoid that reacts with an alkene (or alkyne) to form a cyclopropane. It is named after Howard Ensign Simmons, Jr. and Ronald D. Smith. It uses a methylene free radical intermediate that is delivered to both carbons of the alkene simultaneously, therefore the configuration of the double bond is preserved in the product and the reaction is stereospecific.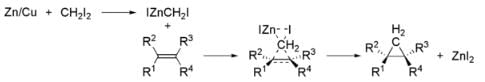 Thus, cyclohexene, diiodomethane, and a zinc-copper couple (as iodomethylzinc iodide, ICH2ZnI) yield norcarane (bicyclo[4.1.0]heptane).
Thus, cyclohexene, diiodomethane, and a zinc-copper couple (as iodomethylzinc iodide, ICH2ZnI) yield norcarane (bicyclo[4.1.0]heptane).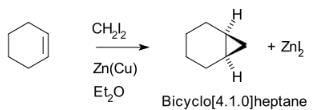
The Simmons–Smith reaction is generally preferred over other methods of cyclopropanation, however it can be expensive due to the high cost of diiodomethane. Modifications involving cheaper alternatives have been developed, such as dibromomethane or diazomethane and zinc iodide. The reactivity of the system can also be increased by using the Furukawa modification, exchanging the zinc‑copper couple for diethylzinc.
The Simmons–Smith reaction is generally subject to steric effects, and thus cyclopropanation usually takes place on the less hindered face. However, when a hydroxy substituent is present in the substrate in proximity to the double bond, the zinc coordinates with the hydroxy substituent, directing cyclopropanation cis to the hydroxyl group (which may not correspond to cyclopropanation of the sterically most accessible face of the double bond): An interactive 3D model of this reaction can be seen at ChemTube3D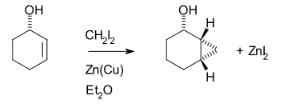
Asymmetric Simmons–Smith reaction:
Although asymmetric cyclopropanation methods based on diazo compounds (see bisoxazoline ligand) exist since 1966, the asymmetric Simmons–Smith reaction was introduced in 1992 with a reaction of cinnamyl alcohol with diethylzinc, diiodomethane and a chiral disulfonamide in dichloromethane: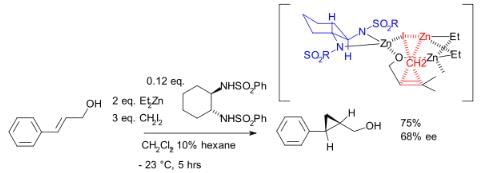 The hydroxyl group is a prerequisite serving as an anchor for zinc. An interactive 3D model of a similar reaction. In another version of this reaction the ligand is based on salen and Lewis acid DIBAL is added:
The hydroxyl group is a prerequisite serving as an anchor for zinc. An interactive 3D model of a similar reaction. In another version of this reaction the ligand is based on salen and Lewis acid DIBAL is added: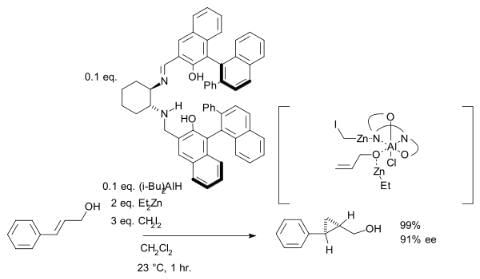
Scope and Limitations:
Achiral Alkenes
The Simmons–Smith reaction can be used to cyclopropanate simple alkenes without complications. Unfunctionalized achiral alkenes are best cyclopropanated with the Furukawa modification (see below), using Et2Zn and CH2I2 in 1, 2-dichloroethane. Cyclopropanation of alkenes activated by electron donating groups proceed rapidly and easily. For example, enol ethers like trimethylsilyloxy-substituted olefins are often used because of the high yields obtained.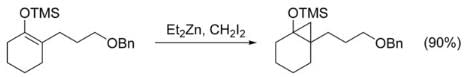 Despite the electron-withdrawing nature of halides, many vinyl halides are also easily cyclopropanated, yielding fluoro-, bromo-, and iodo-substituted cyclopropanes.
Despite the electron-withdrawing nature of halides, many vinyl halides are also easily cyclopropanated, yielding fluoro-, bromo-, and iodo-substituted cyclopropanes.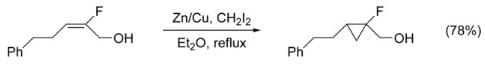 The cyclopropanation of N-substituted alkenes is made complicated by N-alkylation as a competing pathway. This can be circumvented by adding a protecting group to nitrogen, however the addition of electron-withdrawing groups decreases the nucleophilicity of the alkene, lowering yield. The use of highly electrophilic reagents such as CHFI2, in place of CH2I2, has been shown to increase yield in these cases.
The cyclopropanation of N-substituted alkenes is made complicated by N-alkylation as a competing pathway. This can be circumvented by adding a protecting group to nitrogen, however the addition of electron-withdrawing groups decreases the nucleophilicity of the alkene, lowering yield. The use of highly electrophilic reagents such as CHFI2, in place of CH2I2, has been shown to increase yield in these cases.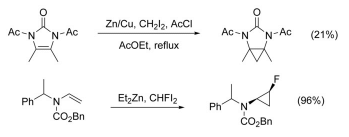
Polyenes
Without the presence of a directing group on the olefin, very little chemoselectivity is observed. However, an alkene which is significantly more nucleophilic than any others will be highly favored. For example, cyclopropanation occurs highly selectively at enol ethers.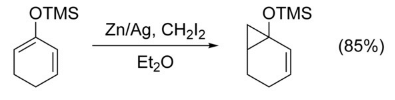
Functional Group Compatibility
An important aspect of the Simmons–Smith reaction that contributes to its wide usage is its ability to be used in the presence of many functional groups. Among others, the haloalkylzinc-mediated reaction is compatible with alkynes, alcohols, ethers, aldehydes, ketones, carboxylic acids and derivatives, carbonates, sulfones, sulfonates, silanes, and stannanes. However, some side reactions are commonly observed.
Most side reactions occur due to the Lewis-acidity of the byproduct, ZnI2. In reactions that produce acid-sensitive products, excess Et2Zn can be added to scavenge the ZnI2 that is formed, forming the less acidic EtZnI. The reaction can also be quenched with pyridine, which will scavenge ZnI2 and excess reagents.
Methylation of heteroatoms is also observed in the Simmons–Smith reaction due to the electrophilicity of the zinc carbenoids. For example, the use of excess reagent for long reaction times almost always leads to the methylation of alcohols. Furthermore, Et2Zn and CH2I2 react with allylic thioethers to generate sulfur ylides, which can subsequently undergo a 2, 3-sigmatropic rearrangement, and will not cyclopropanate an alkene in the same molecule unless excess Simmons–Smith reagent is used.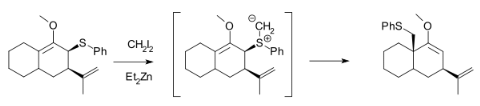
Uses in synthesis:
Most modern applications of the Simmons–Smith reaction use the Furukawa modification. Especially relevant and reliable applications are listed below.
Insertion to form γ-keto esters
A Furukawa-modified Simmons-Smith generated cyclopropane intermediate is formed in the synthesis of γ-keto esters from β-keto esters. The Simmons-Smith reagent binds first to the carbonyl group and subsequently to the α-carbon of the pseudo-enol that the first reaction forms. This second reagent forms the cyclopropyl intermediate which rapidly fragments into the product.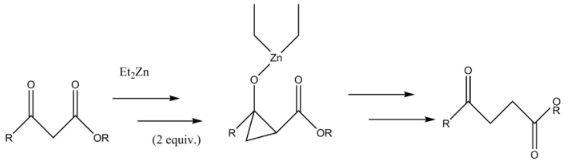
Formation of amido-spiro [2.2] pentanes from allenamides
A Furukawa-modified Simmons–Smith reaction cyclopropanates both double bonds in an allenamide to form amido-spiro [2.2] pentanes, featuring two cyclopropyl rings which share one carbon. The product of monocyclopropanation is also formed.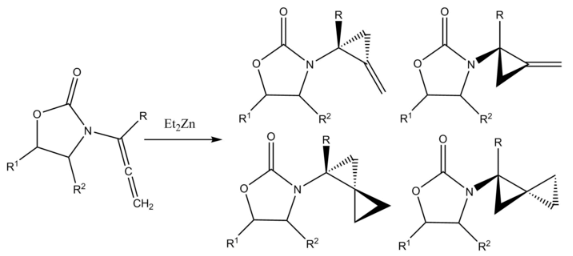
Natural Products Synthesis
Cyclopropanation reactions in natural products synthesis have been reviewed. The β-lactamase inhibitor Cilastatin provides an instructive example of Simmons-Smith reactivity in natural products synthesis. An allyl substituent on the starting material is Simmons-Smith cyclopropanated, and the carboxylic acid is subsequently deprotected via ozonolysis to form the precursor.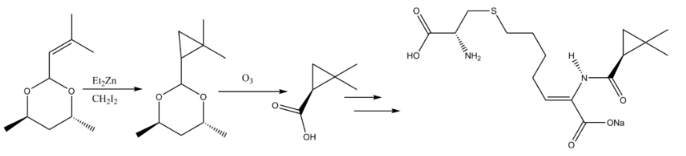
|
44 videos|102 docs|52 tests
|
FAQs on Organic Reactions With Mechanism and Applications (Part -2) - Organic Chemistry
| 1. What is the mechanism of an organic reaction? |  |
| 2. What are some common organic reactions with their applications? | |
| 3. How can the mechanism of an organic reaction be determined? | |
| 4. What are the applications of understanding organic reaction mechanisms? | |
| 5. What are the challenges in studying organic reaction mechanisms? | |



video lectures
,Free
,Objective type Questions
,Extra Questions
,Sample Paper
,Organic Reactions With Mechanism and Applications (Part -2) | Organic Chemistry
,past year papers
,Previous Year Questions with Solutions
,Organic Reactions With Mechanism and Applications (Part -2) | Organic Chemistry
,Semester Notes
,Exam
,shortcuts and tricks
,MCQs
,study material
,ppt
,Important questions
,Summary
,mock tests for examination
,Viva Questions
,practice quizzes
,Organic Reactions With Mechanism and Applications (Part -2) | Organic Chemistry
;


Organic Reactions With Mechanism and Applications (Part -2) Free PDF Download
Importance of Organic Reactions With Mechanism and Applications (Part -2)
Organic Reactions With Mechanism and Applications (Part -2) Notes
Organic Reactions With Mechanism and Applications (Part -2) Chemistry Questions
Study Organic Reactions With Mechanism and Applications (Part -2) on the App
|
© EduRev
|
Education Revolution
|



|





