Organic Reactions with Mechanism (Part -2) | Organic Chemistry PDF Download

TYPE OF ALDOL REACTIONS
1. Intramolecualr
2. Intermolecular
3. Mixed Aldol
4. Retro Aldol
The intramolecular aldol condensation
The intramolecular aldol condensation is a powerful tool for obtaining five- and six-member rings. This is an important step in the Robinson annulation reaction.
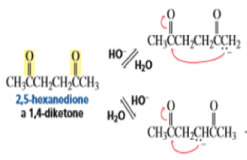
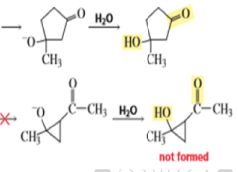
Intramolecular Aldol Additions Because a 1,4-diketone has two different sets of two different intramolecular addition products can potentially form—one with a five-membered ring, the other with a three-membered ring. The greater stability of five- and six-membered rings causes them to be formed preferentially (Section 2.11). In fact, the fivemembered ring product is the only product formed from the intramolecular aldol addition of a 1,4-diketone.
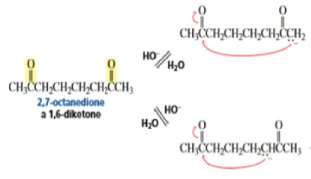
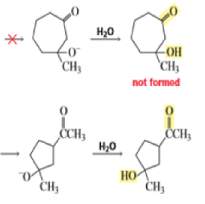
The intramolecular aldol addition of a 1,6-diketone potentially can lead to either a seven- or a five-membered ring product. Again, the more stable product—the one with the five-membered ring—is the only product of the reaction.
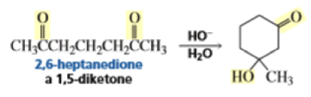
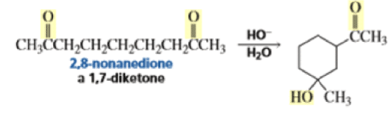
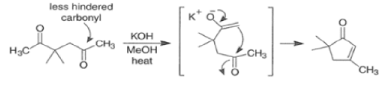
In the following base-catalyzed intramolecular aldol reactions, both enolates are formed reversibly. However, cyclization is faster via attack of the enolate at the less hindered carbonyl group.
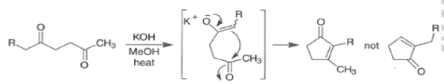
With 1,5-dicarbonyl compounds, two modes of ring closure are often possible. In the example shown below, the more stable (higher-substituted) enone is formed preferentially (thermodynamic c~ntrol).~’ The observed distribution of products is the result of equilibration via retro-nldol reaction
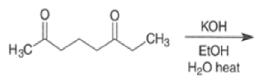
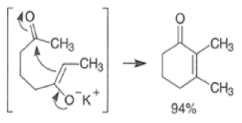
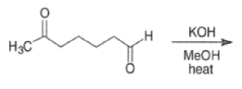
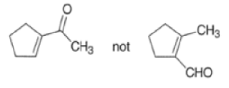
Intramolecular aldol reactions of 1,6-diketones or 1,6-keto-aldehydes afford the corresponding cyclopentenyl carbonyl compound.
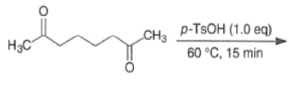
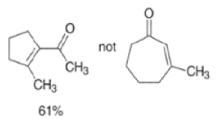
MIXED ALDOL REACTIONS
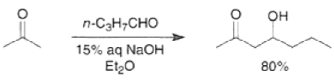
Aldol reactions between two different carbonyl compounds are called mixed or crossed aldol reactions. With aqueous bases, these reactions are of little synthetic value if both reactants have a-hydrogens because they afford mixtures of products. However, under carefully controlled conditions, it is possible to condense ketones with aldehydes in the presence of dilute sodium hydroxide to furnish ²hydroxy ketones, provided that the aldehyde is added slowly to the ketone.
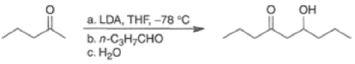
Changing from thermodynamic to kinetic conditions (LDA), it is possible to carry out crossed aldol reactions with good control, as exemplified below.
CANNIZZARO REACTIONI
Non-enolisable aldehydes or aldehydes without a-hydrogen atoms undergo disproportionation in presence of strong base. This is called Cannizzaro reaction.
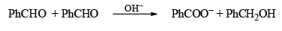
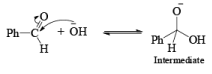
1. When base is not in excess:
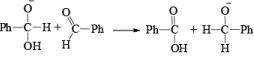
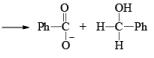
i.e., the overall reaction is 3rd order (2nd order w.r.t aldehyde and 1st order w.r.t base).
2. When base is in excess:
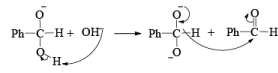
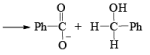
BAEYER-VILLIGER OXIDATION [BVO]
It tolerates with α,βunsaturated ketones, the oxidation with peroxyacids generally occurs at the carbonyl group and not at the C=C double bond; 2) the regiochemistry depends on the migratory aptitude of different alkyl groups.
For acyclic compounds, R’ must usually be secondary, tertiary, or vinylic. For unsymmetrical ketones the approximate order of migration is tertiary alkyl > secondary alkyl > aryl > primary alkyl > methyl 3) the rearrangement step occurs with retention of the stereochemistry at the migrating center; 4) a wide variety of peroxyacids as oxidizing agents can be used to perform the BVO and their activity is ranked as follows: CF3CO3H > monopermaleic acid > monoperphthalic acid > 3,5-dinitroperbenzoic acid > p-nitroperbenzoic acid > mCPBA ~ performic acid > perbenzoic acid > peracetic acid » H2O2 > t-BuOOH. The great synthetic utility of the reaction is derived from its stereospecificity and often high degree of regioselectivity.
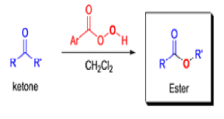
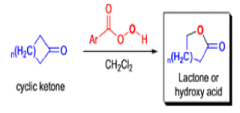
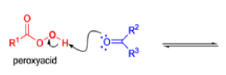
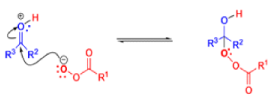
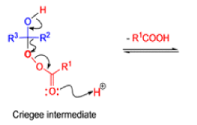

The rearrangement proceeds in a concerted manner that’s why it is stereospecific. Thus, a chiral migrating group maintains its chiral integrity in the product. The overall reaction represents an insertion of oxygen between the carbonyl carbon and the migrating group.
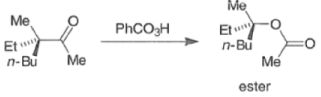
In addition to electronic factors, steric and conformational constraints as well as reaction conditions may influence the ease of migration. However, the regiochemistry can usually be controlled by proper choice of migrating group.
For example, oxidation of methyl ketones results almost exclusively in the formation of acetates. Thus, the BaeyerVilliger oxidation is not only stereospecific but frequently regioselective.
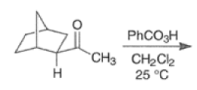
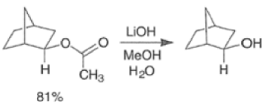
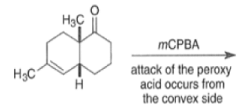
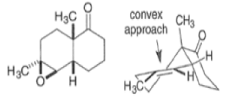
By controlling reaction conditions and by proper choice of the peroxy acid, it is often possible to favor the BaeyerVilliger reaction over epoxidation. An illustrative example of the usefulness of the Baeyer-Villiger reaction is the stereospecific, and regio- and chemoselective conversion of the unsaturated bicyclic ketone sliown below to a cyclopentene containing three consecutive stereogenic centers.
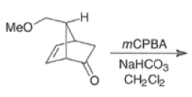
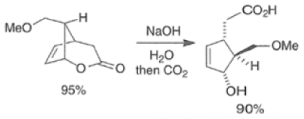
Epoxidation of the electron-deficient double bond in a, P-unsaturated ketones may be complicated by the BaeyerVilliger reaction, an oxidation involving the carbonyl group
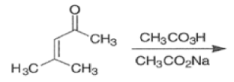
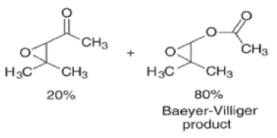
However, if the double bond and the carbonyl group are not conjugated, the former generally reacts faster with peroxy acids than the carbonyl group.
|
44 videos|102 docs|52 tests
|
FAQs on Organic Reactions with Mechanism (Part -2) - Organic Chemistry
| 1. What is an organic reaction? |  |
| 2. What is a reaction mechanism? | |
| 3. How are organic reactions classified? | |
| 4. What is the significance of understanding reaction mechanisms in organic chemistry? | |
| 5. How can reaction mechanisms be determined experimentally? | |




Objective type Questions
,Viva Questions
,Semester Notes
,Summary
,shortcuts and tricks
,ppt
,mock tests for examination
,Previous Year Questions with Solutions
,Free
,study material
,Organic Reactions with Mechanism (Part -2) | Organic Chemistry
,practice quizzes
,video lectures
,past year papers
,MCQs
,Sample Paper
,Organic Reactions with Mechanism (Part -2) | Organic Chemistry
,Organic Reactions with Mechanism (Part -2) | Organic Chemistry
,Important questions
,Extra Questions
,Exam
;


Organic Reactions with Mechanism (Part -2) Free PDF Download
Importance of Organic Reactions with Mechanism (Part -2)
Organic Reactions with Mechanism (Part -2) Notes
Organic Reactions with Mechanism (Part -2) Chemistry Questions
Study Organic Reactions with Mechanism (Part -2) on the App
|
© EduRev
|
Education Revolution
|



|






within 7 days!