Organic Reactions with Mechanism (Part - 3) | Organic Chemistry PDF Download

DAKIN OXIDATION
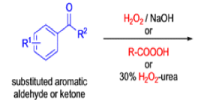
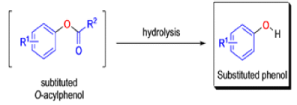
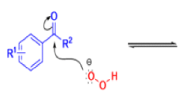
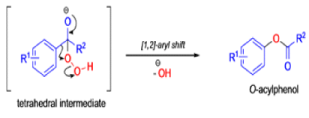
Aliphatic aldehydes are smoothly oxidized to carboxylic acids when treated with organic peracids (RCO3H) or hydrogen peroxide (H2O2). Aromatic aldehydes, however, undergo a more complex reaction in which the aldehyde group is converted to the acylated phenolic hydroxyl group. The reaction works best if the aromatic aldehyde or ketone is electron-rich (-R, -OH, -OR, -NH2, or -NHR substituents in the ortho or para positions). When the aromatic ring is substituted with electron-withdrawing groups, the product of the oxidation is usually carboxylic acid. The Dakin oxidation is usually performed using the following reagents: alkaline H2O2, acidic H2O2, peroxybenzoic acid, peroxyacetic acid.
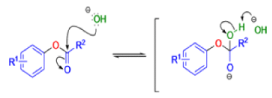
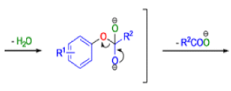
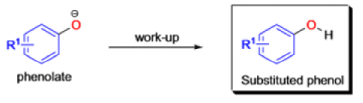
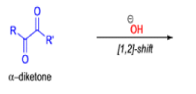
BENZILIC ACID REARRANGEMENT
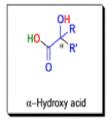
αdiketones rearrange to give salts of αhydroxy acids upon treatment with base (e.g. NaOH), This process is called the benzilic acid rearrangement. The reaction takes place with both aliphatic and aromatic αdiketones and αketo aldehydes. Usually diaryl diketones undergo benzilic acid rearrangements in excellent yields, but aliphatic α-diketones that have enolizable αprotons give low yields due to competing aldol condensation reactions.
Cyclic α-diketones undergo the synthetically useful ring-contraction benzilic acid rearrangement reaction under these conditions. When alkoxides or amide anions are used in place of hydroxides, the corresponding esters and amides are formed. This process is called the benzilic ester rearrangement. Alkoxides that are readily oxidized such as ethoxide (EtO–) or isopropoxide (Me2HCO–) are not synthetically useful, since these species reduce the αdiketones to the corresponding αhydroxy ketones. Aryl groups tend to migrate more rapidly than alkyl groups. When two different aryl groups are available, the major product usually results from migration of the aromatic ring with the more powerful electron-withdrawing group(s).
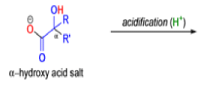
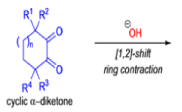
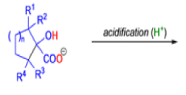
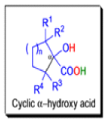
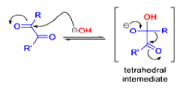
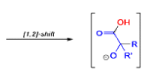
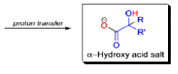
The benzilic acid rearrangement is an irreversible process. The first step of the mechanism is the addition of the nucleophile (HO–, alkoxide, or amide ion) across the C=O bond to give a tetrahedral intermediate. The next step is aryl or alkyl migration to form the corresponding αhydroxy acid salt.
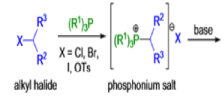
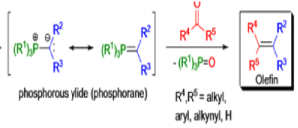
WITTIG REACTION
The formation of carbon-carbon double bonds from carbonyl compounds and phosphoranes(phosphorous ylides) is known as the Wittig reaction. The active reagent in this transformation is the phosphorous ylide, which is usually prepared from a triaryl- or trialkylphosphine and an alkyl halide (1° or 2°) followed by deprotonation with a suitable base (e.g., RLi, NaH, NaOR, etc.), solvents such as THF, Et2O, or toluene are used.. The general features of the Wittig reaction are: 1) the phosphonium salts are usually prepared using triphenylphosphine, and the phosphorous ylides are generated before the reaction or in situ) the ylides are water as well as oxygen-sensitive; 3) the phosphorous ylides chemoselectively react with aldehydes (fast) and ketones (slow), other carbonyl groups (e.g., esters, amides) remain intact during the reaction; 4) the stereoselectivity, E-or Z-selectivity, is influenced by many factors such as type of ylide, There are two different types of ylides depending on the nature of the R2 and R3 substituents: 1) Stabilized Ylides : In the “stabilized” ylides the alkyl halide component has at least one strong electron-withdrawing group (—COOR, —SOOR, etc.), which stabilizes the formal negative charge on the carbon; 2) “nonstabilized” ylides usually have only alkyl substituents, aldehydes afford olefins with high (Z)-selectivity; 6) “stabilized” ylides give predominantly (E)-olefins with aldehydes.
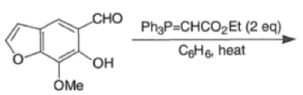
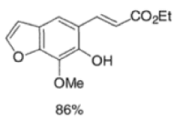
Example
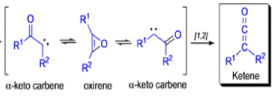
WOLFF REARRANGEMENT
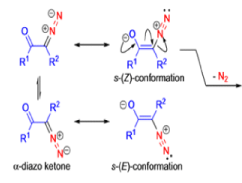
The conversion of αdiazo ketones into ketenes and products derived from ketenes is known as the Wolff rearrangement. In 1902, L. Wolff was studying the chemistry of αdiazo ketones when he observed that upon treatment with silver oxide and water, diazoacetophenone rearranged to give phenylacetic acid. When the reaction medium contained aqueous ammonia, phenylacetamide was formed.
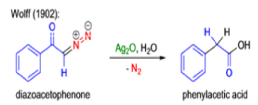
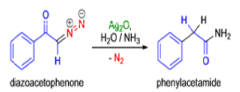
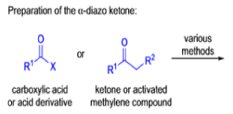
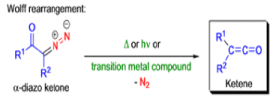
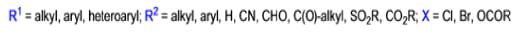
Mechanism
THE PINACOL REARRANGEMENT
When the 1,2-diol ‘pinacol’ is treated with acid, a rearrangement takes place.
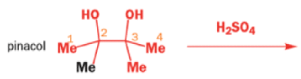
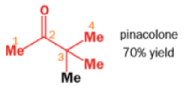
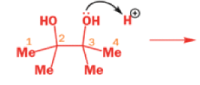
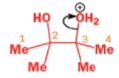
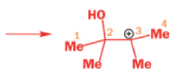
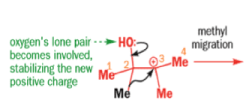
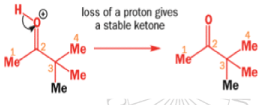
e.g.
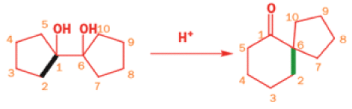
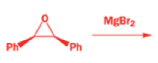
Epoxides rearrange with Lewis acids in a pinacol fashion
The intermediate cation in a pinacol rearrangement can equally well be formed from an epoxide, and treating epoxides with acid, including Lewis acids such as MgBr2, promotes the same type of reaction.

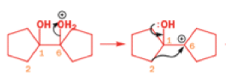
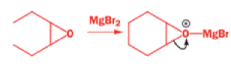
Rearrangement of epoxides with magnesium salts means that opening epoxides with Grignard reagents can give surprising results.


The alkyllithium reaction is quite straightforward as long as the alkyllithium is free of lithium salts. A clue to what has happened with the Grignard reagents comes from the fact that treating this epoxide with just MgBr2 (no RMgBr) gives an aldehyde.

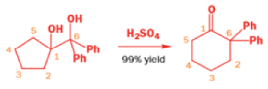
With a Grignard reagent, rearrangement occurs faster than addition to the epoxide, and then the Grignard reagent adds to the aldehyde.
Some pinacol rearrangements have a choice of migrating group
When an unsymmetrical diol or epoxide rearranges, it is important which way the reaction goes. Usually, the reaction leaves behind the more stable cation.
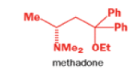
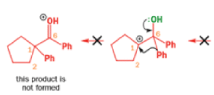
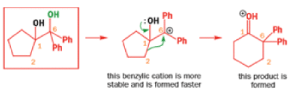
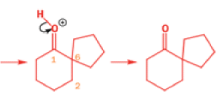
Semipinacol rearrangements are pinacol reactions with no choice about which way to go
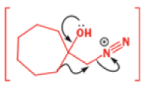
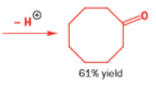
In 1971, French chemists needed this seven-membered cyclic ketone. A reasonable starting material to use is this diol, because it can be made in two steps from the natural product isonopinone.
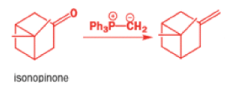
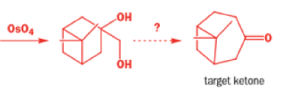
The reaction they needed for the last stage is a pinacol rearrangement—the primary hydroxyl group needs persuading to leave as the ring expands. The problem is, of course, that the tertiary hydroxyl group is much more likely to leave since it leaves behind a more stable carbocation.
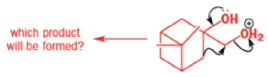

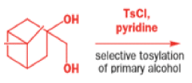
The solution to this problem is to force the primary hydroxyl group to be the leaving group by making it into a tosylate. The primary hydroxyl group reacts more rapidly with TsCl than the tertiary one because it is less hindered.
A weak base is now all that is needed to make the compound rearrange in what is known as a semipinacol rearrangement.
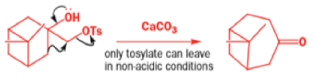
Semipinacol rearrangements are rearrangements in which a hydroxyl group provides the electrons to ‘push’ the migrating group across, but the ‘pull’ comes from the departure of leaving groups other than water—tosylate in this example, but typically also halide or nitrogen (N2). Since tosylation occurs at the lesshindered hydroxyl group of a diol, not only can semipinacol rearrangements be more regioselective than pinacol rearrangements, but their regioselectivity may be in the opposite direction.
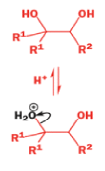

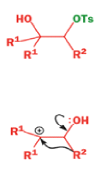
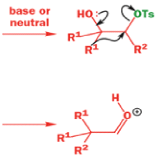
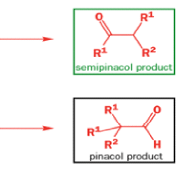
Semipinacol rearrangements of diazonium salts
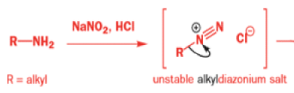
Aromatic amines can be converted to diazonium salts by treatment with acidic sodium nitrite.

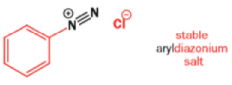
Aryldiazonium salts are stable but alkyldiazonium salts are not: nitrogen gas is the world’s best leaving group, and, when it goes, it leaves behind a carbocation.
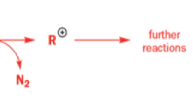
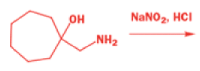
One of the ‘further reactions’ this carbocation can undergo is rearrangement. If the starting amine is a 2-amino alcohol, the cation can be stabilized by a semipinacol rearrangement.
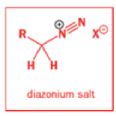
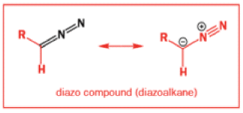
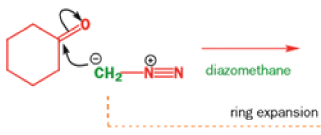
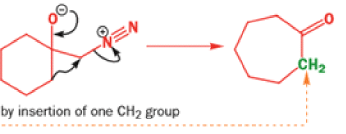
While alkyldiazonium salts are unstable, their conjugate bases, diazoalkanes, are stable enough to be prepared and are nucleophilic towards carbonyl compounds. Diazoalkanes are neutral compounds having one fewer proton than diazonium salts and are delocalized structures with a central sp nitrogen atom.
When diazomethane (a compound we will investigate in more detail in Chapter 40) adds to a ketone, the product undergoes a ring expansion by rearrangement of the same type of intermediate.
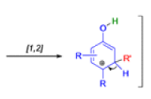
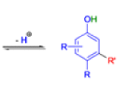
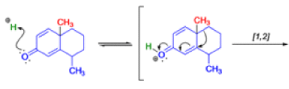
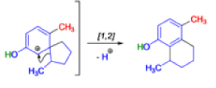
THE DIENONE–PHENOL REARRANGEMENT
The acid- and base-catalyzed or photochemically-induced migration of alkyl groups in cyclohexadienones isknown as the dienone-phenol rearrangement, and is widely used for the preparation of highly substituted phenols.
Dienone-phenol rearrangements require only moderately strong acidic media (e.g., H2SO4 in acetic acid, acetic anhydride, Lewis acidic clay), and they are considerably exothermic due to the formation of very stable aromatic compounds.
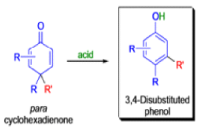
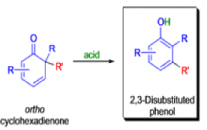
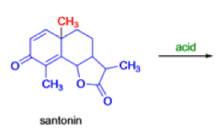
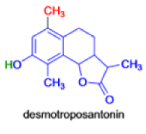
In 1893, A. Andreocci described the rearrangement of santonin to desmotroposantonin upon acidic treatment, butit was only in 1930 that the starting material and the product of this rearrangement were carefully characterized.
Oestrone lacks one of progesterone’s methyl groups, probably removed in the body as CO2 after oxidation., a man whose work led directly to the invention of the contraceptive pill, showed that another derivative of cholesterol could be rearranged to the oestrone analogue 1-methyloestradiol—notice how the methyl group has this time migrated to an adjacent carbon atom.
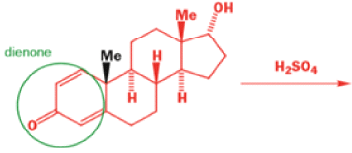
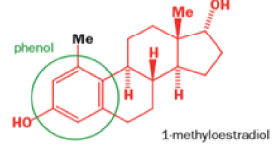
Mechanism

HOFMANN REARRANGEMENT
The conversion of primary carboxamides to the corresponding one-carbon shorter amines is known as the Hofmann rearrangement (also known as the Hofmann reaction).
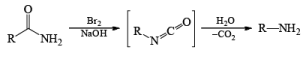
Mechanism
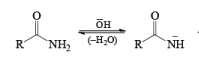
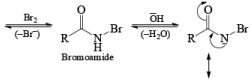
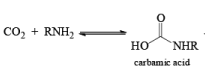
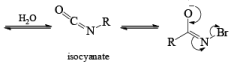
LOSSEN REARRANGEMENT
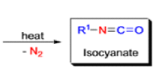
The lossen rearrangement is the conversionof a hydroxamic acid to an isocyaniate via the formation of an O-acyl, sulphonyl, or phosphoryl intermediate hydroxamic acid O-derivative and then conversion to its conjugate base.
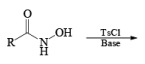
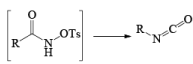
The isocynate can be used further to generate ureas in the presence of amines or generate amines in the presence of
H2O
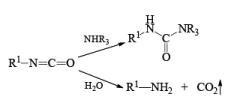
CURTIUS REARRANGEMENT
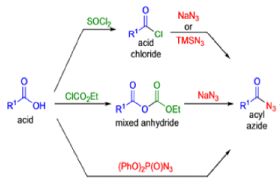
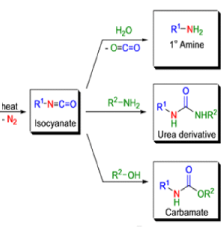
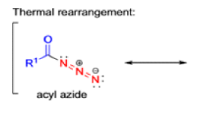
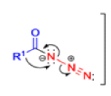
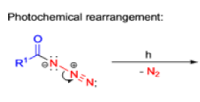
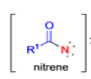
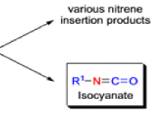
The thermal decomposition (pyrolysis) of acyl azides to the corresponding isocyanates is known as the Curtius rearrangement. The rearrangement is catalyzed by both protic acid and Lewis acids and the decomposition temperature is significantly lowered compared to the uncatalyzed reaction. Acyl azides can be prepared in several different ways: 1) reacting acid chlorides or mixed anhydrides with alkali azide or trimethylsilyl azide 2) treating acylhydrazines with nitrous acid or nitrosonium tetrafluoroborate; 3) treating carboxylic acids with diphenyl phosphoryl azide (DPPA). The product isocyanate can be isolated if the pyrolysis is conducted in the absence of nucleophilic solvents. If the reaction is carried out in the presence of water, amines (R-NH2), or alcohols (R-OH), the corresponding amines, ureas, and carbamates are formed. The Curtius rearrangement is a very general reaction and can be applied to carboxylic acids containing a wide range of functional groups. It is also possible to induce a Curtius rearrangement under photochemical conditions, but this pathway gives rise to several side-products in addition to the desired isocyanate.
Mechanism
|
44 videos|102 docs|52 tests
|
FAQs on Organic Reactions with Mechanism (Part - 3) - Organic Chemistry
| 1. What are organic reactions? |  |
| 2. What is a reaction mechanism? | |
| 3. How are organic reactions classified? | |
| 4. What is the importance of understanding organic reaction mechanisms? | |
| 5. How can one determine the mechanism of an organic reaction? | |




shortcuts and tricks
,Exam
,video lectures
,mock tests for examination
,Objective type Questions
,Organic Reactions with Mechanism (Part - 3) | Organic Chemistry
,Summary
,Organic Reactions with Mechanism (Part - 3) | Organic Chemistry
,Viva Questions
,Organic Reactions with Mechanism (Part - 3) | Organic Chemistry
,Previous Year Questions with Solutions
,past year papers
,Free
,Important questions
,Semester Notes
,Sample Paper
,ppt
,practice quizzes
,MCQs
,study material
,Extra Questions
;






Organic Reactions with Mechanism (Part - 3) Free PDF Download
Importance of Organic Reactions with Mechanism (Part - 3)
Organic Reactions with Mechanism (Part - 3) Notes
Organic Reactions with Mechanism (Part - 3) Chemistry Questions
Study Organic Reactions with Mechanism (Part - 3) on the App
|
© EduRev
|
Education Revolution
|



|





