Organic Reactions with Mechanism (Part - 5) | Organic Chemistry PDF Download

SIMMONS-SMITH REACTION
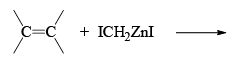
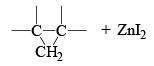
The Simmous-Smith reagent, named for the two DuPont chemists who discovered it, is made by adding methylene iodide to the “zinc-copper couple” (zinc dust that has been activated with an impurity to cooper). The reagent probably resembles iodomethyl zinc iodide, ICH2ZnI. This kind of reagent is called a carbenoid because it reacts much like a carbene, but it does not actually contain a divalent carbon atom.
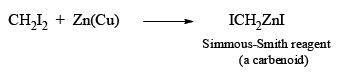
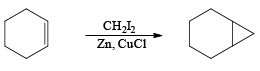
e.g.
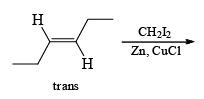
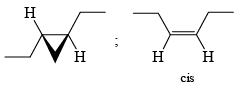
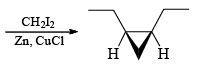
DIELS-ALDER REACTION
The discovery of the [4n + 2n] cycloaddition reaction by Otto Diels (Nobel Prize, 1950) and Kurt Alder (Nobel Prize, 1950), a landmark in synthetic organic chemistry, permits the regio- and stereoselective preparation of both carbocyclic and heterocyclic ring systems. Its application can result simultaneously in an increase of (I) the number of rings, (2) the number of asymmetric centers, and (3) the number of functional groups. The reaction controls the relative stereochemistry at four contiguous centers. The Diels-Alder reaction can be depicted as a concerted n4% nh (suprafacial) cycloaddition. While depicted as a concerted process, the reaction has been proposed to proceed in a nonsynchronous manner via an unsymmetrical transition state.30 Cycloadditions are controlled by orbital symmetry (Woodward-Hoffman rules) and can take place only if the symmetry of all reactant molecular orbitals is the same as the symmetry of the product molecular orbital. Thus, an analysis of all reactant and product orbitals is required. A useful simplification is to consider only the frontier molecular orbitals.” These orbitals are the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUNIO). The orbital symmetry must be such that bonding overlap of the terminal lobes can occur with suprafacial geometry; that is, both new bonds are formed using the same face of the diene.

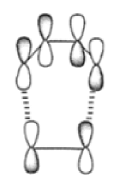
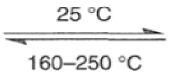
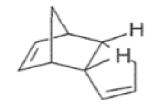
The Diels-Alder reaction is reversible, and many adducts, particularly those formed from cyclic dienes, dissociate into their components at higher temperature. Indeed, a retro-Diels-Alder reaction is the principal method for preparing cyclopentadiene prior to its use in cycloaddition reaction.

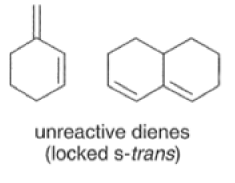
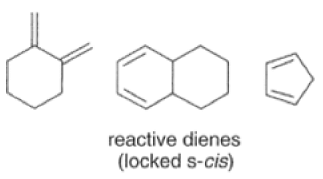
Dienes The diene must be able to adopt the s-cis geometry (s refers to the single bond) in order for cycloaddition to occur. Thus, mono-substituted (E)-dienes are more reactive than (Z)-dienes since (E)-dienes more readily adopt the reactive s-cis information.
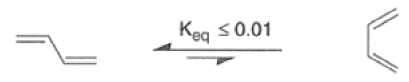

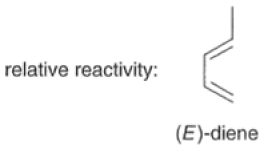
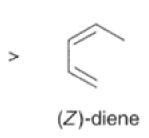
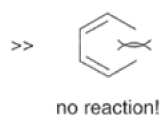
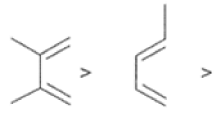
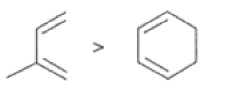
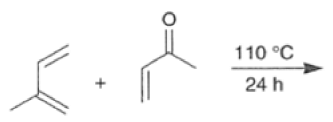
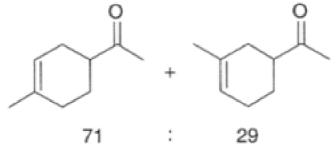
In electron-demand Diels-Alder reactions, dienes are activated by electron-donating substituents, such as alkyl, -NR,, and -OR. Electron-rich dienes accelerate the reac- tion with electron-deficient dienophiles, as illustrated by the relative reactivity trend shown below.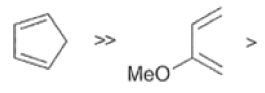
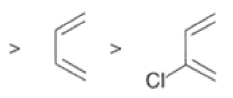
Dienophiles Dienophiles are activated by electron-withdrawing substit~ents.~~ ~lk~l groups, by means of inductive electron donation and steric effects, tend to reduce the rate of cycloaddition.
Relative dienophile reactivity
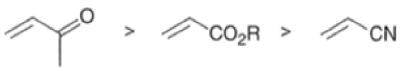
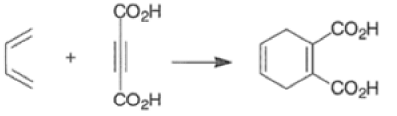
The reactivity of dienophiles may be increased by conjugation with additional electron-withdrawing groups. Doubly activated alkynes and 1,4-benzoquinones are particularly good participants in the Diels-Alder reaction.
Relative dienophile reactivity
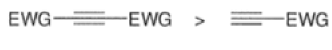

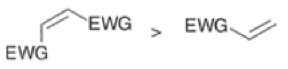

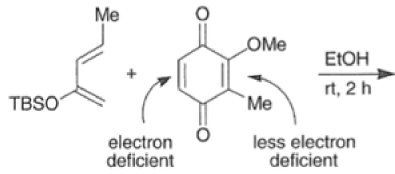
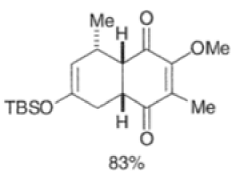
In situ-generated cyclopentadienones are among the most reactive dienophile~.~’ The cyclopentadienone moiety is generated by eliminative processes in the presence of a diene, as shown below.42 The doubly activated alkene reacts in preference to the singly activated one.
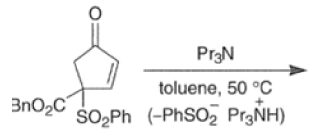
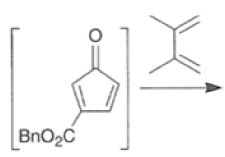
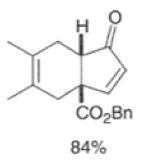
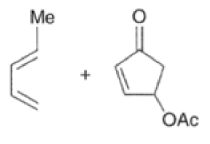
Cyclopentadienone equivalents, such as 4-acetoxy-2-cyclopenten- 1 -one, below, may serve as conjunctive reagents for tandem Diels-Alder reactions providing poly- cyclic adducts.

Lewis Acid Activation
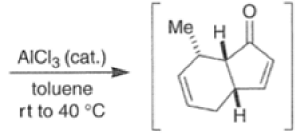
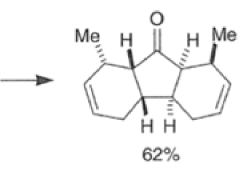
Many Diels-Alder reactions are accelerated by Lewis acid catalysts” such as BF3.OEt2, AlCl3 SnCl4, TiCl3.
These increase the rate of reaction by complexation with conjugated C=O and C=N groups in the diene.
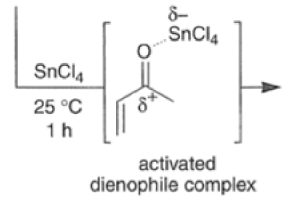

The reaction selectivity is often improved when using Lewis acids
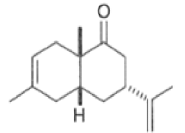
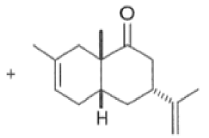

Regiochemistry
The regioselectivity of Diels-Alder reactions ranges from moderate to very high.53 AS shown below in Tables, the cycloaddition reactions of monosubstituted dienes proceed with good electivity. Generally, the more powerful the electronic effect of the diene substituent is, the more regioselective is the reaction.
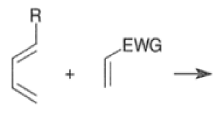
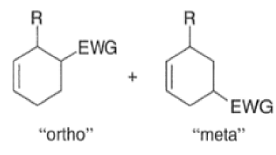



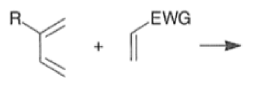
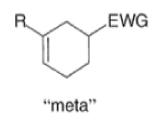
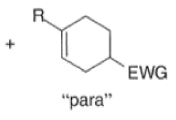



A “simplistic” approach to predicting the regiochemical course of a Diels- Alder reaction is to consider the polarization of the diene and of the dienophile by examining the resonance forms, and then join the atoms with unlike charges to form a six-member ring, as exemplified below. However, this approach fails to account for some reactions that occur with good regiochemistry


To rigorously predict the regiochemistry of Diels-Alder reactions, one has to apply frontier molecular orbital theory.s6
* Estimate the energies of the HOMO and the LUMO of both components.
Identify which HOMO-LUMO pair is closer in energy.
* Using this HOMO-LUMO pair, estimate the relative sizes of the coefficients of the atomic orbitals on the atoms at which bonding is to occur.
* Match up the larger coefficient of one component with the larger on the other.
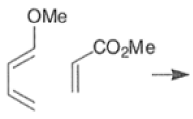
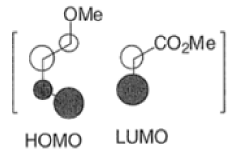
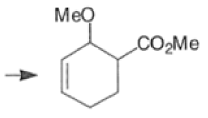
Note: In the example above, the HOMO of the diene possessing an electron-donating group (OMe) has the largest coefficient at the terminus of that group, and the LUMO has the larger coefficient at the end of the electronwithdrawing COOMe group.
Stereochemistry
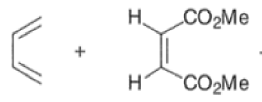
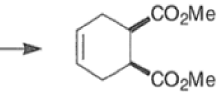
An important aspect of the Diels-Alder reaction is its stereospecificity, wherein the relative stereochemical relationships present in the starting materials are preserved throughout the course of the reaction.
The stereochemistry of both the diene and dienophile are retained in the adduct. Note that the initial suprafacial cycloadduct formed may in some cases be prone to isomerization.
Suprafacial with respect to diene:
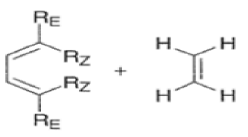
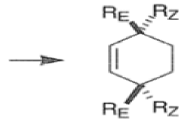
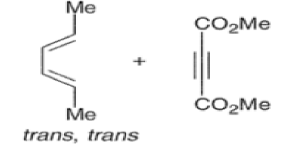
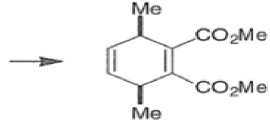
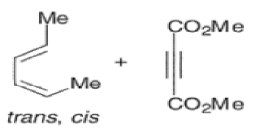
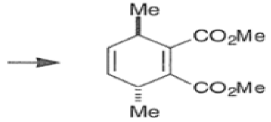
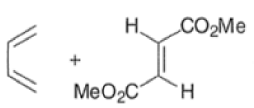
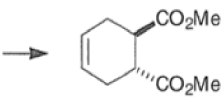
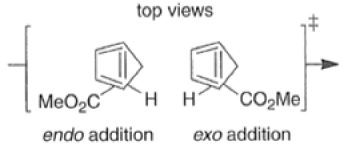
endo-Principle. The dienophile can undergo reaction via two different orientations with respect to the plane of the diene (endo or exo). Maximum overlap of the n-orbitals of the diene and dienophile favors formation of the endoadduct (secondary orbital overlap between the dienophile activating substituent and the diene) exo-Transition state (—COOMe away from n-system of diene)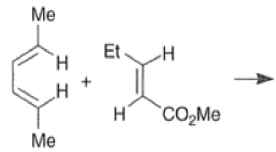
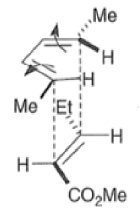
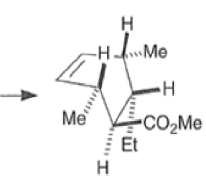
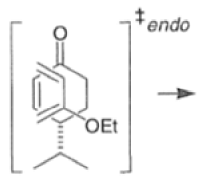
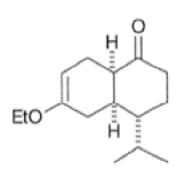
Note that the endo-product has the electron-withdrawing group cis to the (E, E)- substituents of the diene. Only one mode of stacking of the reagents is shown, with the diene above the dienophile. The other possibility is with the diene below the dienophile, leading to the enantiomer. In general, endo-selectivity is determined by a balance of electronic and steric effects. The endo-selectivity may be improved by using Lewis acid catalysis to lower the temperature required for cycloaddition, as shown below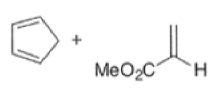
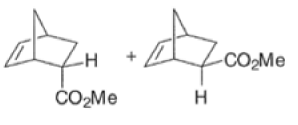
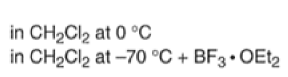

The diene will approach the dienophile from the face opposite a bulky substituent, and a dienophile will approach the less hindered face of the diene.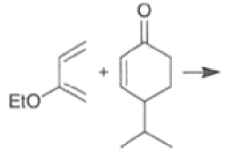

BECKMANN REARRANGMENT
Ketooximes when heated with certain inorganic reagents undergo rearrangement to form amides. This is known as Beckmann rearrangement.
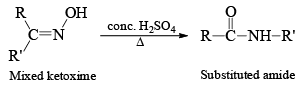
The reagents most commonly used are concentrated sulhuric acid, phosphorus pentachloirde, polyphoshoric acid, thionyl chloride, liquid sulphur dioxide and Benzenesulphonyl chloride (C6H5SO2Cl).
Mechanism
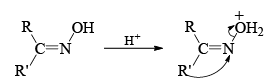
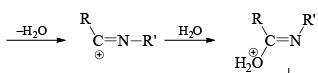
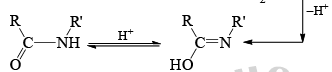
Stereochemistry ¨
- Which groups migrate in the Beckmann rearrangement? For a mixed ketomixes, the answer really matters.
Studies have shown that the group placed anti to the —OH migrates. - The migrating group may be an alkyl or aryl. However, hydrogen does not migrate. That's why aldoximes never give unsubstituted amide.
- The stereochemistry of the reaction shows that the arrangement is concerted with the departure of the leaving group. The reaction is stereospecific. However when the catalyst is a Bronsted acid, migration is not stereospecific. Probably, under these conditions the syn and anti forms are interconvertible.
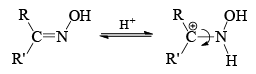
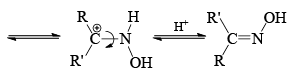
Now we can rewrite the sentence about the migrating group in a bit different way. The group anti to the —OH migrates in Backmann rearrangement under non-isomerizing conditions. When the migrating group is an electron releasing group, the rate of the reaction has been found to be accelerated. This clearly implies that the migration of the group is the slow step. Of course, all Beckmann rearrangement needed no necessarily have the same ratedetermining step.
Very close to Backmann rearrangement is Beckmann fragementation.
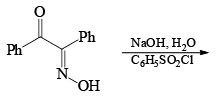
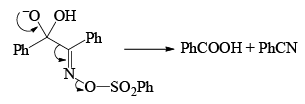
|
44 videos|102 docs|52 tests
|
FAQs on Organic Reactions with Mechanism (Part - 5) - Organic Chemistry
| 1. What is an organic reaction? |  |
| 2. What is a reaction mechanism? | |
| 3. What are some common types of organic reactions? | |
| 4. How are reaction mechanisms determined? | |
| 5. Why are reaction mechanisms important in organic chemistry? | |



video lectures
,past year papers
,Organic Reactions with Mechanism (Part - 5) | Organic Chemistry
,shortcuts and tricks
,Summary
,Previous Year Questions with Solutions
,Free
,Organic Reactions with Mechanism (Part - 5) | Organic Chemistry
,Exam
,Viva Questions
,Semester Notes
,study material
,Organic Reactions with Mechanism (Part - 5) | Organic Chemistry
,MCQs
,Objective type Questions
,Extra Questions
,practice quizzes
,Sample Paper
,Important questions
,ppt
,mock tests for examination
;


Organic Reactions with Mechanism (Part - 5) Free PDF Download
Importance of Organic Reactions with Mechanism (Part - 5)
Organic Reactions with Mechanism (Part - 5) Notes
Organic Reactions with Mechanism (Part - 5) Chemistry Questions
Study Organic Reactions with Mechanism (Part - 5) on the App
|
© EduRev
|
Education Revolution
|



|






within 7 days!