Splitting Of D Orbital Energies In Fields Of Other Symmetry - Coordination Chemistry | Inorganic Chemistry PDF Download

Splitting of d Orbital Energies in Fields of Other Symmetry
Tetrahedral Complexes: In tetrahedral complexes, none of the ligands approach directly any of the d orbitals. However, the ligands come closer to t he orbitals directed to edges of the cube (i.e. dxy, dyz and dzx) than those directed to the centres of the cube (dx2-y2 and dz2).
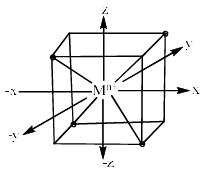
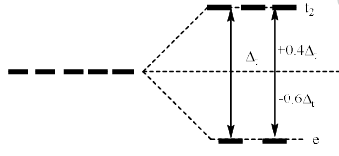
Therefore, the dxy, dyz and dzx orbitals of metal cation experience more repulsion and of higher energy than t hose of dx2 – y2 and dz2 orbitals. After splitting, the dxy. dyz and dzx destabilize (t2 set) b y a factor of 0.4 ∆t and the dx2 – y2 and dz2 orbitals (e set) are stabilized by 0.6 ∆t. The difference in energy of t2 and e set is denoted as ∆t where t stands for tetrahedral. The crystal field splitting in tetrahedral complexes is shown below.
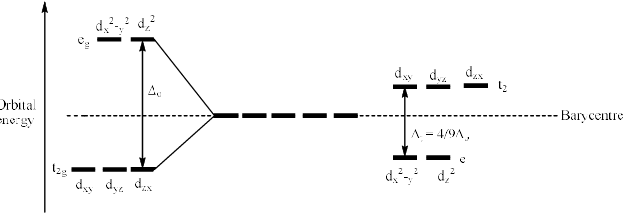
Since ∆t is significantly smaller than ∆o, tetrahedral complexes are high-spin. Also, since smaller amounts of energy are needed for a t2 to e transition (tetrahedral) than for an eg to t2g transition (octahedral), corresponding octahedral and tetrahedral complexes often have different colours.
Tetrahedral complexes are always high spin because:
(a) ∆t = 4/9 ∆0 i.e. ∆t is much smaller than ∆0.
(b) ∆t is alwa ys much smaller than the pairing energy.
Due to these reasons, no pairing occurs in d3, d4, d5, d6, and d7 tetrahedral complexes. Therefore, tetrahedral complexes are always high spin irrespective of strong or weak ligands.
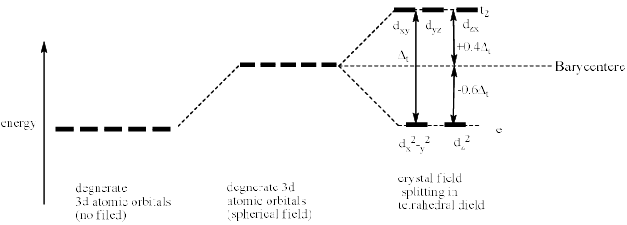
The crystal field splitting in tetrahedral complexes is smaller than that in octahedral complexes. It is observed that
∆t = 4/9 ∆0
(a) In octahedral there are six ligands whereas in tetrahedral low ligands come to interact with metal ions. Hence the repulsion decreases by a factor of 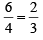
(b) In octahedral, all the six ligands come along the axes while in tetrahedral they come between the axes. Therefore, again repulsion is decreases by a factor of
To these two factors, the crystal field splitting decreases I tetrahedral by a factor of 4/9 than in octahedral i.e.
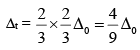
CFSE in Tetrahedral Complexes
In tetrahedral CFSE = (no. of e–s in e orbital) × (–0.6 ∆t) + (no. of e-s in t2 orbital) × (–0.4 ∆t)
or CFSE = ne × (–0.6 ∆t) + nt2 × (+0.4∆t)
In terms of ∆o
CFSE = [ne × (–0.6) + nt2 × (+0.4 )] 4/9 ∆o
CFSE = [–0.27∆o × ne + 0.18∆o × nt2]
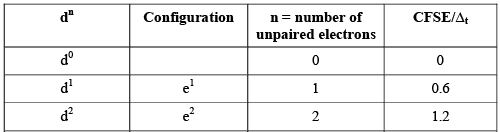
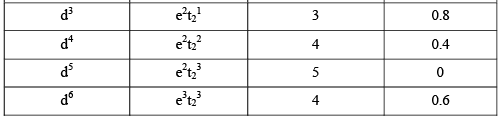
The CFSE values for various configurations of tetrahedral complexes are given in the following table.
Square Planar Complexes
A square planar arrangement of ligands can be formally derived from an octahedral array by removal of two trans ligands.
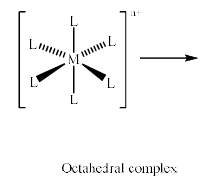
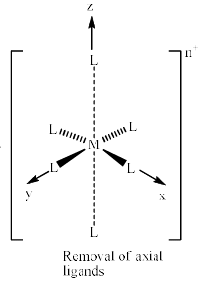
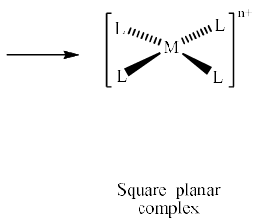
If we remove the ligands lying along the z axis, then the dz2 orbital is greatly stabilized; the energies of the dyz and dxz orbitals are also lowered, although to a smaller extent. The resultant ordering of the metal d orbitals is shown below:
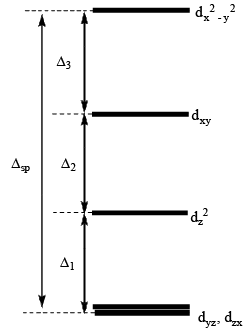
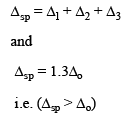
The fact that square planar d8 complexes such as [Ni(CN)4]2– are diamagnetic is a consequence of the relatively large energy difference between the dxy and dx2 – y2 orbitals.
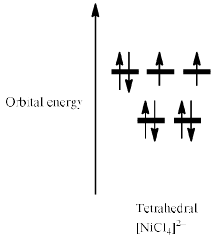
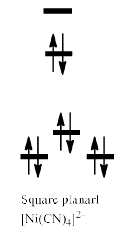
Thus, [NiCl4]2– is paramagnetic while [Ni(CN)4]2– is diamagnetic. Alt hough [NiCl4]2– is tetrahedral and paramagnetic, [PdCl4]2– and [PtCl4]2– are square planar and diamagnetic. This difference is a consequence of the larger crystal field splitting observed for second and third row metal ions compared with their first row congener; Pd(II) and Pt(II) complexes are invariably square planar.
Other Crystal Fields
Crystal field splittings for some common geometries with the relative splittings of the d orbitals with respect to ∆o is given below. But this comparison is only valid for MLx type complexes containing like ligands, and so only applies to simple complexes.
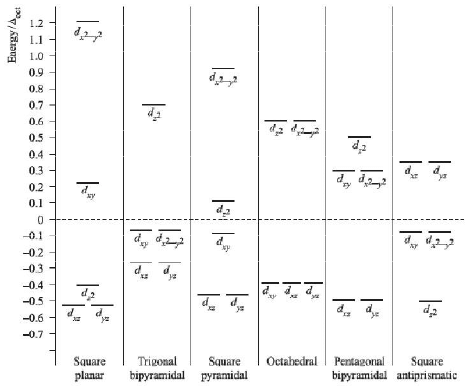
|
50 videos|92 docs|41 tests
|
FAQs on Splitting Of D Orbital Energies In Fields Of Other Symmetry - Coordination Chemistry - Inorganic Chemistry
| 1. What is the splitting of d orbital energies in fields of other symmetry? |  |
| 2. How does the symmetry of the ligand field affect the splitting of d orbital energies? | |
| 3. How can the splitting of d orbital energies be experimentally determined? | |
| 4. What factors influence the magnitude of the splitting of d orbital energies? | |
| 5. How does the splitting of d orbital energies affect the properties of coordination complexes? | |



Splitting Of D Orbital Energies In Fields Of Other Symmetry - Coordination Chemistry | Inorganic Chemistry
,video lectures
,Splitting Of D Orbital Energies In Fields Of Other Symmetry - Coordination Chemistry | Inorganic Chemistry
,Summary
,Viva Questions
,Splitting Of D Orbital Energies In Fields Of Other Symmetry - Coordination Chemistry | Inorganic Chemistry
,mock tests for examination
,practice quizzes
,MCQs
,Semester Notes
,ppt
,Objective type Questions
,Previous Year Questions with Solutions
,Free
,Exam
,Important questions
,study material
,Extra Questions
,past year papers
,Sample Paper
,shortcuts and tricks
;


Splitting Of D Orbital Energies In Fields Of Other Symmetry - Coordination Chemistry Free PDF Download
Importance of Splitting Of D Orbital Energies In Fields Of Other Symmetry - Coordination Chemistry
Splitting Of D Orbital Energies In Fields Of Other Symmetry - Coordination Chemistry Notes
Splitting Of D Orbital Energies In Fields Of Other Symmetry - Coordination Chemistry Chemistry Questions
Study Splitting Of D Orbital Energies In Fields Of Other Symmetry - Coordination Chemistry on the App
|
© EduRev
|
Education Revolution
|



|





