Partition Functions and Their Relation to Thermodynamic Quantities - Physical Chemistry, CSIR-NET - Government Jobs PDF Download

Molecular Partition Functions
Introduction
We have been introduced to the three main ensembles used in statistical mechanics and some examples of calculations of partition functions were also given. In chemistry, we are concerned with a collection of molecules. If the molecules are reasonably far apart as in the case of a dilute gas, we can approximately treat the system as an ideal gas system and ignore the intermolecular forces. The present chapter deals with systems in which intermolecular interactions are ignored. The next chapters will include detailed consideration of intermolecular forces. In ensemble theory, we are concerned with the ensemble probability density, i.e., the fraction of members of the ensemble possessing certain characteristics such as a total energy E, volume V, number of particles N or a given chemical potential μ and so on. The molecular partition function enables us to calculate the probability of finding a collection of molecules with a given energy in a system. The equivalence of the ensemble approach and a molecular approach may be easily realized if we treat part of the molecular system to be in equilibrium with the rest of it and consider the probability distribution of molecules in this subsystem (which is actually quite large compared to systems containing a small number of molecules of the order of tens or hundreds). Since we are dealing with number of particles of the order of Avogadro number, the ensemble description and the molecular descriptions are equivalent. The energies of atoms and molecules are quantized. While atoms have only electronic energy levels, molecules have quantized energy levels arising from electronic, vibrational and rotational motion. A schematic energy level diagram is shown in Fig. 3.1.
We have already seen that in the canonical ensemble, the probability of a system having energy Ei is proportional to the Boltzmann factor and is given in terms of the canonical
partition function q by
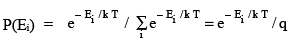 (3.1)
(3.1)
Where q is defined by
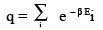 (3.2)
(3.2)
Here, b = 1/ kT and e– βEi is called the Boltzmann factor. Often the Boltzmann constant is written as kB. But when there is no ambiguity, we will simply write k. Once we know the probability distribution for energy, we can calculate thermodynamic properties like the energy, entropy, free energies and heat capacities, which are all average quantities. To calculate P(Ei)s we need the energy levels of a system. The energy ("levels") of a system can be built up from the molecular energy levels. We will consider
the simpler problem of molecular energy levels which are pictorially shown in the Fig 3.1.

Figure 3.1 A schematic diagram showing electronic (bold lines), vibrational and rotational energy levels. The electronic quantum umbers are shown to the extreme right.
Vibrational quantum numbers are to shown in the extreme left. The rotational quantum numbers are shown between the vibrational levels.
Electronic, Vibrational, Rotational and Translational Partition Functions
The electronic energy levels are generally very widely separated in energy compared to the thermal energy kT at room temperature. In each electronic level, there are several
vibrational levels and for each vibrational level, there are several rotational states. This is a simplified and useful model to start with. The total energy is a sum of all these energies and is given by
Etotal = Eelectronic + Evibrational + Erotational + Etranslational + Eothers (3.3)
The term Eothers includes nuclear spin energy levels and may also be used later to include the interactions between the first four. Assuming the first three to be independent and neglecting the last term, the molecular partition function (ie, a sum over the molecular energy states) is given by
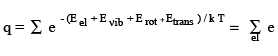
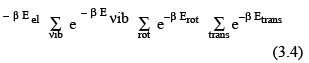
Here, the summation is over the electronic, vibrational and rotational states can be done separately since they are assumed to be independent. Therefore,
q = q el q vib q rot q trans (3.5)
The molecular partition q function is written as the product of electronic, vibrational, rotational and partition functions.
The partition function is a sum over states (of course with the Boltzmann factor b multiplying the energy in the exponent) and is a number. Larger the value of q, larger the
number of states which are available for the molecular system to occupy. Since Eel > Evib > Erot > Etrans, there are far too many translational states available compared to
the rotational, vibrational and electronic states. qel is very nearly unity, qvib and qrot are in the range of 1 to 100 while q trans can be much in excess of 10 20. We shall calculate the values of these qs and indicate how these qs are useful in calculating the equilibrium constants and also in certain cases, the rate constants.
Using the standard formulae for the translational, rotational and vibrational energy levels, we will now calculate the molecular translational, vibrational and rotational
partition functions for diatomic molecules first.
The Translational Partition Function, qtr.
Consider a molecule confined to a cubic box. A molecule inside a cubic box of length L has the translational energy levels given by Etr = h2 (nx2 + ny2 + nz2) / 8 mL2 where nx, ny and nz are the quantum numbers in the three directions (See Chapter 1 for details). The translational partition function is given by
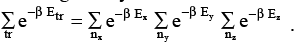 (3.6)
(3.6)
∴q tr = qx qy qz, (3.7)
Which is the product of translational partition functions in the three directions. Since the levels are very closely spaced, we can replace the sum by an integral
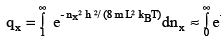
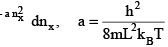 (3.8)
(3.8)
using
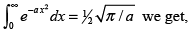
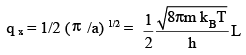
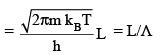 (3.9)
(3.9)
where, Λ is the de Broglie thermal wavelength given by
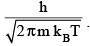
Multipying qx, qy and qz, and using V = volume of the box = L3, we have,
q tr = [ 2 π m kBT / h2]3/2 V = V / Λ3 (3.10))
This is usually a very large number (10 20) for volumes of 1 cm 3 for a typical small molecular mass. This means that such a large number of translational states are accessible
or available for occupation by the molecules of a gas.
Example 3.1 Calculate the translational partition function of an I2 molecule at 300K. Assume V to be 1 liter.
Solution: Mass of I2 is 2 X 127 X 1.6606 X 10-27 kg
2πmkBT = 2 X 3.1415 X (2 X 127 X 1.6606 X 10-27 kg) X 1.3807 X 10-23 J/K X 300 K
= 1.0969 X 10-44 J kg
Λ = h / (2 π m kB T)1/2 = 6.6262 X 10-34 J s / (1.0969 X 10-44 J kg)1/2 = 6.326 X 10-12m
q tr = V / Λ3 = 1000 X 10-6 m3 / (6.326 X 10-12 m )3 = 3.95 X 1030. This means that 3.95 X
1030 quantum states are thermally accessible to the molecular system
The Rotational Partition Function of a Diatomic
The rotational energy levels of a diatomic molecule are given by
 (3.11)
(3.11)
Here, 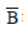 is the rotational constant expresses in cm-1. The rotational energy levels are given by
is the rotational constant expresses in cm-1. The rotational energy levels are given by
by εJ = J(J +1)h2 /8π2I,where I is the moment of inertia of the molecule given by μr2 where, μ is the reduced mass and r, the bond length. Often, the energies are also expressed in terms of the rotational temperature, Θr or Θrot , which is defined as
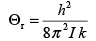 (3.12)
(3.12)
In the summation for the expression for q rot, Eq. (3.13), we can do an explicit summation
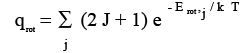 (3.13)
(3.13)
if only a few terms contribute. The factor (2J+1) for each term accounts for the degeneracy of a rotational state J. If energy EJ is degenerate with (2 J + 1) states corresponding to it, then, the Boltzmann factor e - E rot,j / k BT has to be multiplied by (2J+ 1) to account for all these states. If the rotational energy levels are lying very close to one another, we can integrate similar to what we did for q trans above to get
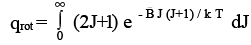 (3.14)
(3.14)
the integration can be easily be done by substituting x = J ( J+1) and dx = (2J + 1) dJ
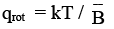 (3.15)
(3.15)
For a homonuclear diatomic molecule, rotating the molecule by 180o brings the molecule into a configuration which is indistinguishable from the original configuration. This leads to an overcounting of the accessible states. To correct for this, we divide the partition function by σ, which is called the symmetry number, which is equal to the distinct number of ways by which a molecule can be brought into identical configurations by rotations. The rotational partition function becomes,
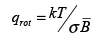 (3.16)
(3.16)
Example 3.2 What is the rotational partition function of H2 at 300K?
Solution: The value of for H2 is 60.864 cm-1. The value of kBT in cm-1 can be obtained by dividing it by hc, i.e., (kBT/hc) = 209.7 cm-1at 300K. σ = 2 for a homonuclear molecule. Therefore, qrot = kT/σ = 209.7/(2X60.864) = 1.723. Since the rotational frequency of H2 is quite large, only the first few rotational states are accessible to at at 300K.
3.1.3 The Vibrational Partition Function of a Diatomic
The vibrational energy levels of a diatomic are given by
En = (n +1/2 ) hν (3.17)
where is ν the vibrational frequency and n is the vibrational quantum number. In this case, it is easy to sum the geometric series shown below
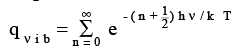 (3.18)
(3.18)
= e - h ν / (2 k BT) (1 + e - h ν / k BT + e - 2 h ν / k BT +.....)
= e − h ν / 2 kT (1 + x + x2 + x3....) (3.19)
= e − h ν / 2 k T [1 / (1- x)] (3.20)
where x = e− h ν/ kBT which is less than 1. Threfore,
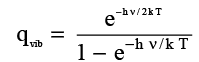 (3.21)
(3.21)
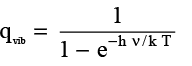 (3.22)
(3.22)
if the zero of energy scale is at hν/2kT . Analogous to Θr, a vibrational temperature Θv or Θvib may be defined as 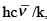 where,
where, 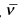 is the vibrational frequency in cm-1.
is the vibrational frequency in cm-1.
Example 3.3 Calculate the vibrational partition function of I2 at 300K.
Solution: The vibrational frequency of of I2 is 214.57 cm-1. hν/kT = 214.57/209.7 = 1.0232
e -hν/kT = 0.3595∴ qvib = 1/(1-0.3595) = 1.561. This implies, as before, that very few vibrational states are accessible.
3.1.4 The Electronic Partition Function
Writing the electronic energy as E1, E2, E3,…with degeneracies g1, g2, g3,…the electronic partition function is given by
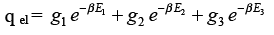 (3.23)
(3.23)
Usually, E1<< E2 or E3. Treating E1 to be the reference value of zero of energy, we get,
q el = g1 (3.24)
which is the ground state degeneracy of the system.
Example 3.4 Find the electronic partition of H2 at 300 K.
Solution: The lowest electronic energy level of H2 is near - 32 eV and the next level is about 5 eV higher. Taking - 32 eV as the zero (or reference value of energy),
qei = e0 + e-5 eV / kT + ...
At 300 K, T = 0.02eV and qel = 1 + e-200 +... ≈ 1.0
Where all terms other than the first are nearly 0. This implies that qel = 1. The physical meaning of this is that only the ground electronic state is generally accessible at room temperature.
Representative data useful for calculating the electronic, vibrational, and rotational partition functions is given in Table 3.1. The electronic degeneracy is represented as g.
The vibrational frequency is written as 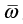 . The dissociation energy is written as D0. It is quite straight forward to calculate the above partition functions for these molecules.
. The dissociation energy is written as D0. It is quite straight forward to calculate the above partition functions for these molecules.
Table 3.1 Representative molecular data for a few diatomics.
|
Molecule |
g |
Bond Length(A) |
|
|
|
|
Force consta nt k (dynes /cm) |
D0 (kcal/mol) |
|
H2 |
1 |
0.7414 |
4400 |
6332 |
60.9 |
87.6 |
5.749 |
103.2 |
|
D2 |
1 |
0.7415 |
3118 |
4487 |
30.45 |
43.8 |
5.77 |
104.6 |
|
N2 |
1 |
1.097 |
2358 |
3393 |
2.001 |
2.99 |
22.94 |
225.1 |
|
O2 |
3 |
1.207 |
1580 |
2274 |
1.446 |
2.08 |
11.76 |
118.0 |
|
Cl2 |
1 |
1.987 |
560 |
805 |
0.244 |
0.351 |
3.2 |
57.1 |
|
CO |
1 |
1.128 |
2170 |
3122 |
1.931 |
2.78 |
19.03 |
255.8 |
|
NO |
2 |
1.15 |
1890 |
2719 |
1.695 |
2.45 |
15.7 |
150.0 |
|
HCl |
1 |
1.275 |
2938 |
4227 |
10.44 |
15.02 |
4.9 |
102.2 |
|
HI |
1 |
1.609 |
2270 |
3266 |
6.46 |
9.06 |
3.0 |
70.5 |
|
Na2 |
1 |
3.096 |
159 |
229 |
0.154 |
0.221 |
0.17 |
17.3 |
|
K2 |
1 |
3.979 |
92.3 |
133 |
0.0561 |
0.081 |
0.10 |
11.8 |
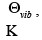
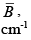
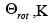
Example 3.5 Express the partition function of a collection of N molecules Q in terms of the molecular partition function q.
Solution: Assuming the N molecules to be independent, the total energy E of molecules is a sum of individual molecular energies Es and

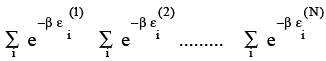 (3.25)
(3.25)
= q.q....q = q N (3.26)
Here 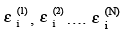 are energies of individual molecules and a sum of all Es can only come from summing over all εi s. Gibbs postulated that
are energies of individual molecules and a sum of all Es can only come from summing over all εi s. Gibbs postulated that
Q = qN / N! (3.27)
The N! in the denominator is due to the indistinguishability of the tiny molecules (or other quantum particles in a collection).
Example 3.6 Derive the Maxwell Boltzmann distribution of molecular speeds.
Solution: If we represent a molecular velocity by vr, it has three independent components vx , vy and vz in the three directions x, y and z. Let us consider monatomic gas of mass m. The probability F ( x , y , z) that given molecule will have velocity components lying between x and x + dx, y and y + dy and z and z + dz can be written as F (x, y, z) dz dy dz = f (x) f (y) f(z) dx dy dz.
F is written as a product of three functions f because x, y and z are independent and since nature does not distinguish between x, y and z (unless directional fields like gravitational or electromagnetic are present), the form of f is the same in the three directions. Again, since there is no distinction between positive and negative x, f depends on | x | or x2. We can rephrase the above equation as
F( ) = f ( ) f ( ) f( ) (3.28)
The only function that satisfies the above equation is an exponential function since 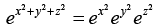 and so we conclude that f ( ) may be written as
and so we conclude that f ( ) may be written as
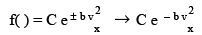 (3.29)
(3.29)
We take only the negative exponent (C and b are positive) because a positive exponent implies that very large velocities have very high probabilities which is highly unlikely.
To evaluate C, We invoke the physical argument that the velocity has to lie somewhere between - ∞ to + ∞ and that the total probability is 1 i.e.,
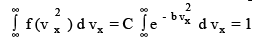
The above integral is a standard integral 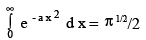
Thus,
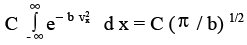 (3.30)
(3.30)
But since we want the right side to be unity, C ( π / b) 1/2 = 1 or C = ( b / π ) 1/2 and 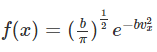
From a probability distribution such as f ( x), average quantities can be determined. The averages of x and x2 are given by
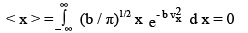 (3.31)
(3.31)
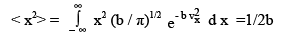 (3.32)
(3.32)
The averages have been denoted by < > . We have also used another standard integral,
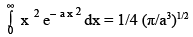 (3.33)
(3.33)
The integral for < x > is zero because the value of the integrand for positive x is equal and opposite to its value at – x. Thus the area on the left of x = 0 is equal and opposite in sign to the area on the right. This is a special case of a general result that the integral of the product of an even function and an odd function of x is zero over a symmetric interval around zero.
To evaluate b, we take the help of the kinetic theory of gases. Do look up the details. The pressure of a gas is given in terms of the mean square velocity (speed) as p = 1/3 (N/ V) m <v2>
Where N/ V = (number of molecules of the gas / volume) = the density of the gas. But N = nNA where n = number of moles and NA , the Avogadro number; and since pV = nRT,
we have
pV = 1/3 Nm < v 2 > = 1/3 n NA m < v2 > = nRT (3.34)
< v2> = 3 RT / mNA = 3 kB T / m (3.35)
Where kB = R / NA is the Boltzmann constant, 1.3806 X 10 -23 J / K. Since <v2> = 3 kB T / m, we have <vx2> = kBT/ m and substituting, we get b = m / 2 kB T. Equations for f and F now become
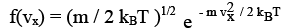 (3.36)
(3.36)
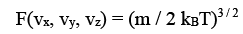
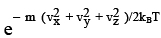 (3.37)
(3.37)
This is the Maxwell - Boltzmann distribution of molecular speeds. F(vx, vy, vz) dvxdvydvz gives the probability of finding an arbitrary molecule with a velocity (vx, vy, vz) in the volume element dvxdvydvz. A more appealing interpretation of the same is that it is the fraction (of the total molecules) of molecules having velocities (vx, vy, vz). Analogous to the radial probability distribution in coordinate space, we can now estimate the probability of finding a particle in a spherical shell of volume 4π v2 dv . This probability in such a spherical shell of radius v and thickness dv is given by
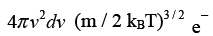
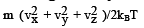 (3.38)
(3.38)
Thermodynamic Functions
We will be restricting ourselves to the canonical ensemble here. Consider a collection of N molecules. The probability of finding a molecule with energy Ei is equal to the fraction of the molecules with energy Ei .
In the collection of N molecules, how many molecules (ni) have the energy Ei ?.This has to be N exp -β Ei / Q. This is because the fraction of molecules ni /N having the energy Ei is e -β Ei / Q which is the same as the probability of finding a molecule with energy Ei in the collection. The average energy is obtaining by multiplying Ei with its probability and summing over all i . ie,
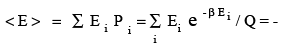
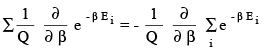
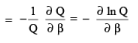 (3.39)
(3.39)
Detailed connection between partition functions and thermodynamic functions has been derived in Chapter 2, Section 2.4.2, The relations between Q and pressure and entropy are given by Eqns. (2.168) and (2.171) respectively. The pressure p can also be obtained as the ensemble average of (-∂E/∂V)T = (-d w/dV)T giving,
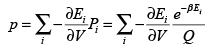
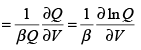 (3.40)
(3.40)
The entropy is given by
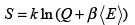 (3.41)
(3.41)
The notation change is that Z is written as Q here and 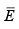 is written as
is written as 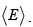
Let us recall the four fundamental thermodynamic relations.
dE = TdS - pdV (3.42)
dH = TdS + Vdp (3.43)
dA = -SdT - pdV (3.44)
dG = -SdT + Vdp (3.45)
Here, E is the internal energy, H = E + pV, the enthalpy, A = E –TS, the Helmholtz free energy and G = H – TS, the Gibbs free energy. Let us now replace <E> by simply E or more correctly by E – E(0), where E(0) is the reference value of E at T = 0. This is necessary because, the absolute value of E is not measurable and only differences can be measured. The same idea holds for H, A as well as G. Therefore, we rewrite the equation for energy as
E – E(0) = - (∂ ln Q / ∂β)V (3.46)
From the equation for enthalpy, H = E + pV, we get,
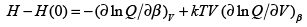 (3.47)
(3.47)
Since p = - (∂A/∂V)T, we get from (3.),
A − A(0) = −kT ln Q (3.48)
And finally we have for G
G − G (0) = −kT ln Q + kTV (∂ ln Q ∂V )β (3.49)
For an ideal gas, pV = nRT where, n = number of moles of the gas and R = NAk = the gas constant and NA is the Avogadro number. Substituting this and Q = qN/N! in the above equation, we get,
G − G(0) = − N kT ln q + kT ln N ! + n RT
Noting that N = nNA and using Stirling’s approximation,
G − G(0) = −nRT ln q + kT ( N ln N − N ) + n RT
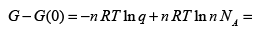
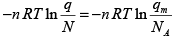 (3.50)
(3.50)
Where, qm = q / n, the molar partition function in units of mol-1 . The function [G(T) –G(0)] / T is referred to as the Gaiuque function.
Example 3.6 The equipartition principle states that each quadratic degree of freedom contributes � kT to the energy at high temperature. Verify this assertion for the rotational, translational and vibrational motions of a diatomic molecule.
Solution
Consider the molecular partition functions. The rotational energy is given by
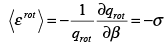
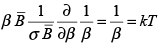 (3.51)
(3.51)
The classical expression for the rotational energy is �I ( ωx2 + ωy2 ) , where I is the moment of inertia and ωx and ωy are the angular velocities in the x and y directions. The rotation along the molecular axis (the z axis here) has no meaning in quantum mechanics because the rotations along the molecular axis lead to configurations which are indistinguishable from the original configuration. The two degrees of freedom have thus given a value of kT. The translational contribution gives,
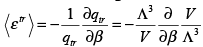
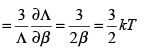 (3.52)
(3.52)
Thus, the three translational degrees of freedom in three dimensions satisfy the equipartition theorem. Turning to the vibrational contribution, we get,
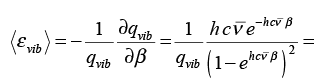
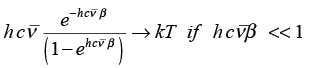 (3.53)
(3.53)
The classical expression for the vibrational energy is � kx2 + � μ vx2. At high temperature the equipartition theorem is valid but at low temperature, only a few vibrational states are occupied and the equipartition principle is not valid.
Example 3.7 Calculate the vibrational heat capacity for diatomic and sketch its temperature dependence.
Solution The vibrational energy is given by the above expression and the heat capacity at constant volume, CV, is given by ∂E/∂T.
We have, ∂ /∂T = (∂β/∂T) (∂ /∂β) = (-1/kT2) (∂ /∂β) = (-k β2) (∂ /∂β) (3.54)
Therefore, CV = (-k β2) (∂ εvib/∂β) =
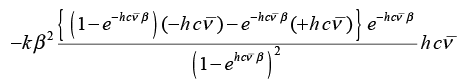
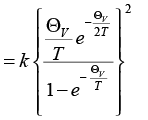 (3.55)
(3.55)
For large T, the molar CV becomes NAk = R and for small T, CV goes to zero as shown in the sketch below. The vibrational temperature ΘV is defined as ΘV = h cν / k. For H2, it has a value of 6323 K and for I2 it is 309 K.
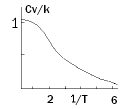
Figure 3.3 Vibrational heat capacity of a diatomic as a function of ΘV /T.
FAQs on Partition Functions and Their Relation to Thermodynamic Quantities - Physical Chemistry, CSIR-NET - Government Jobs
| 1. What is a partition function and how is it related to thermodynamic quantities? |  |
| 2. How does the partition function relate to the internal energy of a system? | |
| 3. How can the partition function be used to calculate the entropy of a system? | |
| 4. What is the significance of the partition function in determining the free energy of a system? | |
| 5. How does the partition function relate to the statistical weight of a system? | |


Extra Questions
,Partition Functions and Their Relation to Thermodynamic Quantities - Physical Chemistry
,Partition Functions and Their Relation to Thermodynamic Quantities - Physical Chemistry
,Partition Functions and Their Relation to Thermodynamic Quantities - Physical Chemistry
,Objective type Questions
,past year papers
,study material
,Viva Questions
,CSIR-NET - Government Jobs
,Free
,Semester Notes
,mock tests for examination
,video lectures
,ppt
,Summary
,Important questions
,MCQs
,CSIR-NET - Government Jobs
,practice quizzes
,Exam
,CSIR-NET - Government Jobs
,Previous Year Questions with Solutions
,Sample Paper
,shortcuts and tricks
;


Partition Functions and Their Relation to Thermodynamic Quantities - Physical Chemistry, CSIR-NET Free PDF Download
Importance of Partition Functions and Their Relation to Thermodynamic Quantities - Physical Chemistry, CSIR-NET
Partition Functions and Their Relation to Thermodynamic Quantities - Physical Chemistry, CSIR-NET Notes
Partition Functions and Their Relation to Thermodynamic Quantities - Physical Chemistry, CSIR-NET Government Jobs Questions
Study Partition Functions and Their Relation to Thermodynamic Quantities - Physical Chemistry, CSIR-NET on the App
|
© EduRev
|
Education Revolution
|



|






within 7 days!