Haemoglobin | Zoology Optional Notes for UPSC PDF Download
Introduction to Hemoglobin
- It is an oxygen/CO2 carrier protein present in the red blood corpuscles of blood. Hemoglobin is a conjugated chromo-protein having heme as its prosthetic group. Heme is the prosthetic group, not only of hemoglobin but also of myoglobin, cytochromes etc.
- Hemoglobin is formed by the combination of heme with globin (protein). Globin is made up of four polypeptide chains (an oligomeric protein). Two of these polypeptides are known as alpha (α) and the other two are known as beta (β). Each alpha chain has 141 amino acids and each beta chain has 146 amino acids, which are arranged in a definite sequence. Its molecular weight is 65,000.
- Due to the characteristic folding of its tertiary structure each polypeptide forms a cup like structure with a pocket like area where the prosthetic group, heme is buried. Heme has iron, which is linked to the imidazole nitrogen of the histidine in positions 58 and 87 of the alpha chains. In the beta chain the heme iron is linked with histidine in positions 92 and 63. Altogether there are four heme groups in one hemoglobin molecule.
Composition of Hemoglobin
Heme:It is an iron-porphyrin compound. It is the prosthetic group embedded in the packet like structure formed by folding of the hemoglobin tertiary structure.
Porphyrin:
Porphyrin is a complex compound with a tetrapyrrole ring structure. Pyrrole is a heterocyclic compound having the following structure.
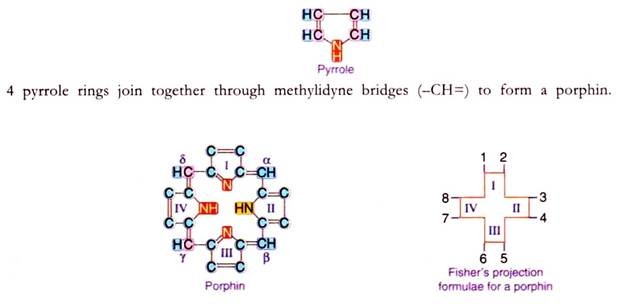
This porphin is substituted by different groups at positions numbered from 1-8 to form the porphyrin. Depending upon the groups (methyl, acetyl, propyl, butyl or venyl) present on these positions different types of porphyrins are identified, that will be seen during the synthesis of heme.
Properties of porphyrins
1. They act both as acids (-COOH) and bases (-NH2).
2. Their isoelectric pH is between 3-4.5.
3. Porphyrins are fluorescent and coloured due to presence of alternating double bonds.
4. Porphyrinogens are colourless.
Biosynthesis of heme
Heme is an iron porphyrin structure, synthesized in the reticuloendothelial cells (bone marrow) of adult human being. Erythropoietin produced in kidney stimulates the formation, maturation and release of erythrocytes by bone marrow.Early stage of erythrocyte cells contain porphyrin, during the course of their development, porphyrin is converted to heme by addition of iron and then to hemoglobin by addition of protein, globin. The type of porphyrin present in heme is protoporphyrin-III (also known as No. IX).
It is synthesized starting from glycine and succinyl-CoA. Given below is the diagrammatic representation of biosynthesis of Heme where ‘A’ stands for acetyl group, ‘P’ stands for propyl group, ‘M’ for methyl group, and ‘V’ for venyl group.
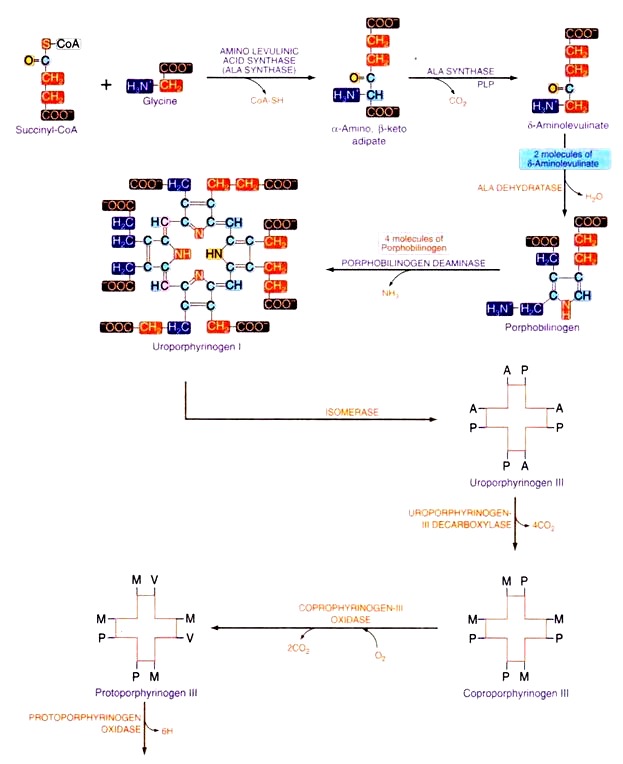
Regulation of heme synthesis
The first enzyme in this sequence i.e. ALA synthase is the key regulatory enzyme for heme synthesis which is inhibited by heme the end product of the metabolism. ALA synthase is a regulatory enzyme. Heme acts as an Apo repressor molecule and is a negative regulator for the synthesis of ALA synthase-I (heme inhibits the gene).Inhibitors of heme synthesis: The following substances inhibit heme synthesis
i. Succinylacetone (SA) is an inhibitor of heme synthesis that acts on the enzyme aminolevulinic acid dehydratase.
ii. N-methyl mesoporphyrin IX blocks iron insertion into protoporphyrin IX and thus acts as an inhibitor of heme synthesis.
iii. Isonicotinic acid hydrazide (INH) is an inhibitor of 6-aminolevulinic acid synthase.
Disorders related to abnormalities in the synthesis of porphyrins
Porphyria’s:
Porphyria’s are a group of diseases in which there is an increased excretion of porphyrins or porphyrin precursors (intermediates of porphyrin synthesis). About 85% of heme synthesis occurs in erythrocyte precursors and 15% in liver. Therefore porphyria’s are classified into two types (1) Erythropoietic and (2) Hepatic. Some of them are inherited while others are acquired.
Hemoglobin Derivatives
There are some derivatives of normal Hb that arise due to metabolic changes in the RBC.
The various hemoglobin derivatives are:
1. Oxyhemoglobin (HbO2):
The main function of hemoglobin is to transport oxygen from the lung to the tissues. In lungs the partial pressure of oxygen is 100 mm of Hg, at this pressure hemoglobin is 95-96% saturated with oxygen. On binding with O2 in the lungs hemoglobin is converted to oxy-hemoglobin (Hb02). O2 is bound to heme iron.
Hb + O2 → HbO2
2. Reduced Hemoglobin (HHb):
Oxy-hemoglobin moves to the tissue where the partial pressure of O2 is 26 mm of Hg due to which oxygen is released into the tissues and in turn H+ binds to Hb and forms reduced hemoglobin.
HbO2 + H+ → HHb + O2
3. Carbaminohemoglobin:
Hemoglobin also binds to CO2 in the tissues. CO2 is bound to the α-amino group at the N-terminal end of each of the four polypeptide chains of hemoglobin to form carbaminohemoglobin. As one CO2 binds O2 is released.
4. Methemoglobin:
In RBC the iron of hemoglobin is normally in ferrous (Fe2+) form, but it is readily oxidized to the ferric (Fe3+) form by hydrogen peroxide formed by RBC cell metabolism, to yield met-hemoglobin. Ferric iron is incapable of binding O2 therefore the functions of hemoglobin are disturbed. Normally 1.7 to 2.4 % of total hemoglobin will be in the form of met-hemoglobin. Increase in the percent of met-hemoglobin is prevented by the peroxidase action of a naturally occurring peptide known as glutathione present in the RBC. Met-hemoglobin is dark brown in colour.
The percent of met-hemoglobin can increase if the person consumes drugs like ferricyanide, nitrite, quinines, hydroxylamine’s, acetanilide and sulfonamide. Higher levels of met-hemoglobin is observed clinically in factory workers who inhale (or contact through skin) aromatic nitro and amino compounds and in patients taking large amounts of acetanilide and sulfonamides. The symptoms are cyanosis (blue skin) and dyspnoea (labored breathing).
Importance of methemoglobin
Met-hemoglobin can be used to overcome cyanide poisoning. By injecting met-hemoglobin it combines with cyanide to form cyanomethemoglobin preventing cyanide poisoning.
5. Carboxyhemoglobin:
Oxy-hemoglobin can bind to carbon monoxide (CO). Even normal, non- oxygenated hemoglobin can bind with CO to form carboxyhemoglobin. [Hb + CO → HbCO]. CO has got an affinity of 200 times more than that of O2 towards Hb. Hemoglobin can bind more readily to CO than to O2. Even if there is a little amount of CO in air, it can displace oxyHb to form carboxyHb. Due to this there will be tissue hypoxia because the oxygen binding capacity is reduced and there is also reduced O2 releasing capacity i.e. it cannot release O2 though it may be bounded to O2.
City dwellers have at least 1% of carboxyhemoglobin which can increase to 8% depending upon the pollution. Over traffic can increase carboxyHb to 40% which leads to death. Clinically such patients show cherry red colour of skin. CO poisoning can be treated if high amount of O2 is provided continuously at high pressure, then at such high concentrations and pressure HbCO is dissociated forming HbO2 + CO. When treatment continues for 2 hours CO is expelled out.
Types of Hemoglobin
There are three types of hemoglobin’s that are normally found in human beings, they are:
HbA;
- Found in normal adult human beings – contains 2α and 2β chains.
HbA2:
- Found in some human beings and is considered normal — contains 2α and 2β chains.
HbF:
- Foetal hemoglobin — found in growing foetus — contains 2α and 2ƴ chains.
Each chain is synthesized by the information obtained from the gene for hemoglobin, α chain is synthesized from a genes of hemoglobin, β chain from β genes of hemoglobin likewise y and 8 from their respective genes. There are 2 pairs of a genes but only one pair each of β, γ and δ genes.
Abnormal hemoglobin’s arise due to mutation in the gene for the hemoglobin synthesis. There are about 300 abnormal hemoglobin’s. Some of them are those which have defect in α genes and some are with defective β chains.
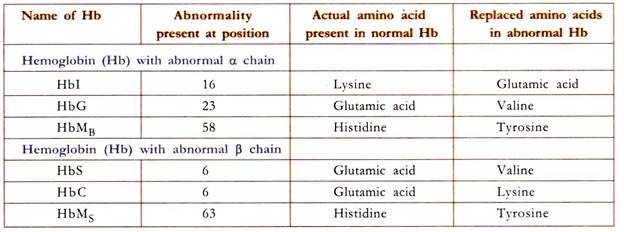
Biochemistry of Abnormality in the Hemoglobin
HbS or Sickle Cell Hemoglobin:
Sickle cell hemoglobin (HbS) arises due to the defect in β chain in which glutamic acid present at the 6th position is replaced by valine. Valine is also present naturally at position one. These two valine residues form hydrophobic interaction producing a sticky patch on HbS. Due to this replacement there is a sticky patch on HbS which appears on the oxy HbS. There is a complementary site to this sticky patch on deoxy HbS and also on deoxy HBA.
The mechanism of biconcave RBC converting to sickle shape is given here under:
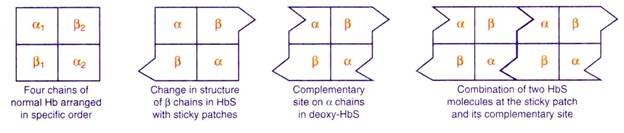
When hemoglobin molecules combine together in chains they form precipitates of HbS. The precipitate formed in the RBC sinks down and the biconcave shape of RBC is converted to sickle shape.
The life span of RBC is reduced to less than half (about 30 days). HbS is very unstable, due to which there is excessive hemolysis. This results in anemia called sickle cell anemia. The physiological changes observed in sickle cell anemia are – physical exertion, weakness, short of breath, leukemia and heart murmurs.
HbM or Methemoglobin:
The defect lies both in α and β chains. This is due to replacement of histidine residue in 58th position in α chain and 63rd position in β chain. Due to this replacement, the iron (Fe) present in the ferrous state is oxidized to ferric state. This ferric iron cannot bind oxygen. Therefore the oxygen carrying capacity is disrupted leading to anemia and hypoxia (low O2 to tissues).
Thalassemia’s:
The defect in thalassemia’s is the decreased rate of synthesis of one of the polypeptide chains of the globin molecule. One of the chains is synthesized in less amounts than the other due to the defect in DNA.
There are two types of thalassemia
- β-thalassemia:
- β-thalassemia occurs due to the decreased synthesis rate of P-chain of globin. Due to the deficiency of P-chain the a-chains either combine among themselves forming a-4-globin or it can combine with γ or δ chains, thereby forming more of HbA2 and HbF. This results in the impairment of the transport of O2 by Hb resulting in hypoxia. There are very low levels of Hb i.e. 2-3 g/100ml (hypochromic cells). The life span of such RBC is greatly reduced. The symptoms include anemia, growth retardation, wasting and fever.
- α-thalassemia:
- α-thalassemia occurs due to the decreased rate of synthesis of α-chain of globin. But this is rarely seen due to the presence of two pairs of genes for a chain in the Hb gene. Due to lack of a chain, the β chain may combine either with δ, γ or among itself forming β4, or β2δ2 or β2γ2.
Ghicose-6-Phosphate Dehydrogenase Deficiency
- It is an X-linked recessive hereditary disease characterised by abnormally low levels of the glucose-6-phosphate dehydrogenase enzyme (abbreviated G6PD or G6PDH). Glucose-6-phosphate dehydrogenase (G6PD) is an enzyme in the pentose phosphate pathway, a metabolic pathway that supplies reducing energy to cells (most notably erythrocytes) by maintaining the level of the co-enzyme nicotinamide adenine dinucleotide phosphate (NADPH).
- The NADPH in turn maintains the level of glutathione in these cells that helps to protect the red blood cells against oxidative damage. G6PD converts glucose-6-phosphate into 6-phosphogluconolactone and is the rate- limiting enzyme of the pentose phosphate pathway.
- Patients with G6PD deficiency are at risk of hemolytic anemia in states of oxidative stress. Individuals with the disease may exhibit non-immune hemolytic anemia in response to a number of causes. This can be due to severe infection, medication and certain foods. Broad beans contain high levels of vicine, divicine, convicine and isouramil—all are oxidants.
- In states of oxidative stress, all remaining glutathione is consumed. Enzymes and other proteins (including hemoglobin) are subsequently damaged by the oxidants, leading to electrolyte imbalance, membrane cross-bonding and phagocytosis and splenic sequestration of red blood cells. The hemoglobin is metabolized to bilirubin (causing jaundice at high concentrations) or excreted directly by the kidney (causing acute renal failure in severe cases).
- Deficiency of G6PD in the alternative pathway causes the build-up of glucose and thus there is an increase of advanced glycation end products (AGE). The deficiency also causes a reduction of NADPH which is necessary for the formation of Nitric Oxide (NO). The high prevalence of diabetes mellitus type 2 and hypertension in Afro-Caribbean’s in the West could be directly related to G6PD deficiency.
- Patients are almost exclusively male, due to the X-linked pattern of inheritance, but female carriers can be clinically affected due to lyonization where random inactivation of an X-chromosome in certain cells creates a population of G6PD deficient red cells coexisting with normal red cells.
- Although female carriers can have a mild form of G6PD deficiency (dependent on the degree of inactivation of the unaffected X- chromosome), homozygous females have been described; in these females there is coincidence of a rare immune disorder termed chronic granulomatous disease (CGD).
G6PD deficiency manifests itself in a number of ways:
- Prolonged neonatal jaundice
- Hemolytic crises in response to:
- Certain drugs
- Certain foods, most notably broad beans
- Illness (severe infections)
- Diabetic ketoacidosis
- Very severe crises can cause acute renal failure
Diagnosis:
The diagnosis is generally suspected when patients from certain ethnic groups develop anemia, jaundice and symptoms of hemolysis after challenge to any of the above causes, especially when there is a positive family history.
Generally, tests will include:
- Complete blood count and reticulocyte count; in active G6PD, Heinz bodies can be seen in red blood cells on a blood film
- Liver enzymes (to exclude other causes of jaundice)
- Lactate dehydrogenase (elevated in hemolysis and a marker of its severity)
- Haptoglobin (decreased in hemolysis)
- A ‘direct anti-globulin test’ (Coombs’ test)—this should be negative, as hemolysis in G6PD is not immune-mediated
- TSH measurement
Pyruvate Kinase Deficiency
- It is an inherited autosomal recessive genetic disorder which affects the survival of red blood cells. Pyruvate kinase deficiency is the second most common cause of enzyme- deficient hemolytic anemia, following G6PD deficiency. A variety of mutations can lead to lowered production, activity, or stability of pyruvate kinase, an enzyme essential to glycolysis. A total lack of this enzyme’s activity will be lethal.
- Because the ability of erythrocytes to manufacture ATP depends on glycolysis, the cells become deficient in energy and hence are unable to maintain the activity of the basolateral Na+/K+-ATPase. This will result in an increase in intracellular [Na+] which will cause water to diffuse passively into the red blood cells (RBC) and will lead to swelling. This swelling will lead to lysis of the RBCs and an increase in plasma bilirubin.
- The increase in plasma bilirubin will lead to jaundice and the lysis of the RBCs will lead to hemolytic anemia. The build-up of reaction intermediates can also increase the level of 2, 3-bisphosphoglycerate (2,3BPG) in the cells and affect tissue oxygenation. This will cause a ‘right shift’ in the hemoglobin oxygen saturation curve, implying a decreased oxygen affinity for the hemoglobin and earlier oxygen unloading than under normal conditions.
- Red blood cells use glycolysis as their sole energy source. In pyruvate kinase deficiency, the last step (phosphoenolpyruvate converted to pyruvate) of glycolysis is unable to occur. A discrepancy between red blood cell energy requirements and ATP generating capacity produces irreversible membrane injury resulting in cellular distortion, rigidity and lysis. This leads to premature erythrocyte destruction by the spleen and liver.
Biosynthesis of Heme and Regulation
Heme, synthesized in the bone marrow's reticuloendothelial cells, involves erythropoietin stimulation. The first enzyme, ALA synthase, is a key regulator inhibited by heme, the end product. Inhibitors such as succinylacetone, N-methyl mesoporphyrin IX, and isonicotinic acid hydrazide can disrupt heme synthesis.
Hemoglobin Derivatives
Metabolic changes in red blood cells lead to various hemoglobin derivatives:
- Oxyhemoglobin (HbO2): Forms in the lungs, binding to oxygen.
- Reduced Hemoglobin (HHb): Releases oxygen in tissues.
- Carbaminohemoglobin: Binds to CO2 in tissues, releasing oxygen.
- Methemoglobin: Results from iron oxidation, disturbing hemoglobin function.
- Carboxyhemoglobin: Forms when hemoglobin binds to carbon monoxide, reducing oxygen-carrying capacity.
Types of Hemoglobin
Three types of hemoglobins are found in humans:
- HbA: Found in normal adults (2α and 2β chains).
- HbA2: Present in some adults (2α and 2β chains).
- HbF: Foetal hemoglobin, found in growing fetuses (2α and 2ƴ chains).
Biochemistry of Abnormalities in Hemoglobin
Several abnormal hemoglobins, arising from gene mutations, lead to conditions such as sickle cell anemia, methemoglobinemia, thalassemias, and G6PD deficiency.
- Sickle Cell Hemoglobin (HbS): Results from a β-chain defect, causing sticky patches and a shift from biconcave to sickle shape, leading to anemia.
- Methemoglobin (HbM): Arises from α and β chain defects, preventing oxygen binding and causing anemia and hypoxia.
- Thalassemias: Result from decreased synthesis of globin chains, impacting oxygen transport and causing anemia.
- G6PD Deficiency: An X-linked recessive condition affecting glucose-6-phosphate dehydrogenase, leading to hemolysis during oxidative stress.
Conclusion
Understanding the intricacies of hemoglobin, its derivatives, types, and abnormalities is crucial for comprehending the intricate processes within red blood cells, influencing oxygen transport and overall health. This comprehensive exploration provides valuable insights into the biochemistry and functions of hemoglobin in health and disease.
|
181 videos|346 docs
|
FAQs on Haemoglobin - Zoology Optional Notes for UPSC
| 1. What is the composition of hemoglobin? |  |
| 2. How is heme synthesized in the body? | |
| 3. What are some substances that inhibit heme synthesis? | |
| 4. What are some disorders related to abnormalities in the synthesis of porphyrins? | |
| 5. Why is methemoglobin important in the body? | |

|
Dec 22, 2024 Last updated |

|
Explore Courses for UPSC exam
|
|
Exam
,Objective type Questions
,past year papers
,Haemoglobin | Zoology Optional Notes for UPSC
,Free
,Semester Notes
,Important questions
,study material
,practice quizzes
,Viva Questions
,mock tests for examination
,Sample Paper
,Previous Year Questions with Solutions
,shortcuts and tricks
,MCQs
,video lectures
,Haemoglobin | Zoology Optional Notes for UPSC
,ppt
,Haemoglobin | Zoology Optional Notes for UPSC
,Summary
,Extra Questions
;






Haemoglobin Free PDF Download
Importance of Haemoglobin
Haemoglobin Notes
Haemoglobin UPSC Questions
Study Haemoglobin on the App
|
© EduRev
|
Education Revolution
|



|




