Rearrangements | Chemistry Optional Notes for UPSC PDF Download
Pinacol Rearrangement
- The pinacol rearrangement is the acid catalyzed rearrangement of a 1,2-diol into a ketone. It is named after the molecule pinacol, pictured below, the simplest substrate to undergo the reaction. One of the alcohols is protonated to make the leaving group (water) while the other OH participates as the rearrangement promoting group.
- After water leaves, generating a tertiary carbocation, the remaining alcohol forms a carbonyl, thus promoting the rearrangement of a methyl group. Deprotonation completes the mechanism to form pincolone and regenerate the catalytic acid.

- For unsymmetrical diols, not surprisingly, you form the most stable carbocation intermediate. You will see an example of this in the problems below. What about substrates where different groups can migrate? The following examples demonstrate that H migrates faster than R, which is consistent with what you saw previously in Intro Orgo.
- An H shift yields a more stable cationic intermediate than if an alkyl group migrates. What about Ph vs H? In this case, the benzene ring migrates faster. These seems strange until we remember our previous chapter on neighboring group participation. A migratory phenyl yields a phenonium ion intermediate that is favored over H migration.

Solved Examples
Example 1: For the following reaction, propose a product and a mechanism to explain its formation.
Ans: Using the same strategy as the pinacol rearrangements above, we quickly generate a tertiary carbocation. Formation of the carbonyl that we know will be in the product promotes rearrangement (migration) of one of the ring bonds. This yields a very interesting result. One of the original 5-membered rings expands to form a new 6-membered ring. So, we can use the pinacol rearrangement as a ring expansion reaction!

Example 2: For the following reaction, propose a product and a mechanism to explain its formation.

Ans: In this example, the two possible carbocations that can form after water leaves are very different. The tertiary carbocation is low enough in energy to form; however, the doubly benzylic, resonance stabilized carbocation is much lower in energy. So, it forms preferentially, resulting in a methyl shift to form the product ketone.

- Another interesting aspect of the pinacol rearrangement is the impact of stereochemistry on the migrating group. If you think about this statement, it seems to contradict what we have shown in all of the reactions up to now in this chapter. How can stereochemistry of the diol (cis or trans) matter when we generate a planar carbon as part of the carbocation intermediate? It turns out, like many reactions in organic chemistry, our first look was an oversimplification. The current understanding is that an achiral carbocation does not form. Instead, a chiral ion pair forms, retaining the stereochemistry of the starting material.
- However, when considering rearrangements of chiral diols, we will sometimes draw the carbocation and sometimes a concerted mechanism depending on what helps most with our understanding. The key is to recognize that what does form is a chiral version of the free carbocation (the ion pair) that promotes the rearrangement. Similar to what you learned in intro orgo for SN2 and E2 reactions, the migrating group must be antiperiplanar to the leaving group. The example and problem below demonstrate that stereochemistry is critically important when considering reactions of cyclic diols.
- We will first consider reaction of the cis cyclohexane diol. Axial leaving groups are much more reactive than equatorial leaving groups, so the reaction occurs with the protonated axial OH leaving accompanied by formation of the new carbonyl and rearrangement of the axial methyl group, the anitperiplanar migrating group. What happens if we run the same reaction on the trans isomer of this diol? See if you can come up with an answer as part of the next problem.

Example 3: What is the product of this reaction? Also, provide a mechanism for its formation.

Ans: Both of the alcohols are equatorial because they are larger than the axial methyl groups and they can form an intramolecular hydrogen bond to stabilize this conformation. So, when a protonated equatorial alcohol leaves, which group is antiperiplanar to participate in the pinacol rearrangement? It's clearly not one of the methyl groups. Thus, the ring bond is the only alternative. As shown in the mechanism, formation of the new carbonyl promotes cleavage of the 5-6 bond to form a new 1-5 bond. The result is a ring contraction from 6- to 5-members! This demonstrates that the pinacol rearrangement can produce both ring contractions and ring expansions, as we saw previously.
- Let's consider one more type of pinacol rearrangement that is very important for complex structures and often appears in total synthesis applications. It is called a semipinacol rearrangement and involves rearrangement under basic conditions where it is impossible to form a carbocation. To understand why this is a key reaction, let's first look at the reaction below. This is a standard pinacol rearrangement.
- As we saw above, with an unsymmetrical diol, we will protonate the tertiary carbocation to form the more stable tertiary carbocation and then do our rearrangement. In this case, a hydrogen shift to yield the ketone product. But, what if we wanted to form the carbocation at the secondary alcohol? It is impossible to do this under acidic conditions.

- The solution is to move away from acidic conditions and think about reactions we know that occur selectively at secondary alcohols. If we can make the secondary alcohol into a good leaving group, we can force that alcohol to leave, reverse the position of the rearrangement promoting O and generate a different product. How do we put that into practice? The reaction sequence below shows the standard way this is accomplished.
- Alcohols react selectively with tosyl chloride from least hindered (primary) to most hindered (tertiary), so we can selectively tosylate the secondary alcohol in the presence of the tertiary alcohol. Next, addition of a base results in deprotonation of the alcohol to form an alkoxide that can promote the rearrangement. Formation of the ketone promotes rearrangement of the ring bond to force out the tosylate leaving group. In this fascinating example, that results in a simultaneous ring expansion and ring contraction!

Payne Rearrangement
- The Payne rearrangement involves reactions of nucleophiles with epoxy alcohols under basic conditions. Like many rearrangements, at first glance it seems confusing. We can rationalize hydroxide adding to an epoxide, but how does the sulfur nucleophile replace the poor OH leaving group? The key here is to think first about deprotonating the primary alcohol and then to focus on neighboring group participation.

- Deprotonating the primary alcohol generates an alkoxide nucleophile that opens the epoxide via an intramolecular SN2 reaction to form a new epoxide. The sulfur nucleophile can now add to the less hindered side of the new epoxide via an intermolecular SN2 reaction. The final protonation step yields the target trans diol.


Benzilic Acid Rearrangement
- The benzilic acid rearrangement involves conversion of a 1,2-diketone into a carboxylic acid. The conditions are deceptively simple, hydroxide followed by an acid quench, and lead to the migration of a benzene ring.
- This mechanism is relatively straightforward. Hydroxide adds to one of the ketones to yield a tetrahedral intermediate. Reforming the carbonyl results in rearrangement of the phenyl onto the second carbonyl. The acid quench ultimately generates the target carboxylic acid.


Favorskii Rearrangement
The Favorskii rearrangement transforms an alpha halo ketone into an ester, as shown in the example below. Upon first inspection, this seems to continue the theme we just saw in the benzilic acid rearrangement. Add the nucleophile to the carbonyl, reform the carbonyl, and have the rearrangement occur pushing out the leaving group. For this substrate, that results in a ring contraction reaction. As we will see in the problems below, this is not the only mechanistic possibility.

Example 4: Propose a mechanism and a product for the following reaction.

Ans: This reaction appears to behave exactly like the reaction shown above. Rearrangement occurs from the tetrahedral intermediate and yields the ester product. See the answers to Exercise #5 for an alternate mechanism to get the same product.

Tiffeneau-Demjanov Rearrangement
- In this rearrangement, a 1,2-aminoalcohol is converted into a ketone as shown in the generic example below. The reagent that promotes this transformation is nitrous (not nitric) acid. If you previously studied nucleophilic aromatic substitution reactions, you might recognize that combining an amine and nitrous acid yields a reactive diazonium intermediate. This is a key step in the Sandmeyer reaction where anilines react with nitrous acid to yield a diazo benzene intermediate that reacts with a variety of nucleophiles to make new benzene derivatives.
- In the Tiffeneau-Demjanov rearrangement, formation of the diazonium intermediate promotes the key rearrangement step. Similar to previous mechanisms, formation of the carbonyl promotes the alkyl shift and loss of the leaving group.
- A historical note about this reaction is to recognize the key role played by Bianka Tchoubar in its development. She conducted the critical experiments in Tiffeneau's lab to determine the scope and limitations of the reaction. Bianka was a key figure in the organic chemistry community in France from the 1930s until the 1980s, resulting in 140 publications.


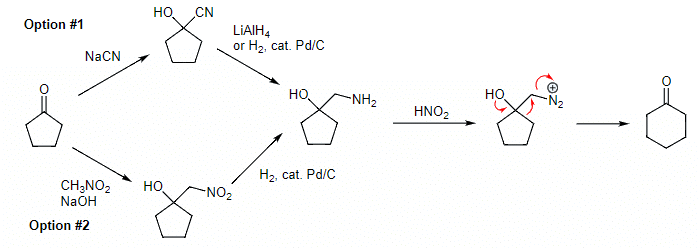
Example 5: First, starting with cyclopentanone, how would you produce the desired aminoalcohol? Second, what product is formed upon exposure of the aminoalcohol to nitrous acid?
Ans: There are two ways to convert cyclopentanone into the target aminoalcohol. In option #1, we add sodium cyanide to yield a cyanohydrin that can be reduced with either lithium aluminum hydride or hydrogen and catalytic palladium. In option #2, we use a Henry reaction to generate a nitroalcohol that can be reduced with hydrogen and catalytic palladium. Adding nitrous acid promotes the Tiffeneau-Demjanov rearrangement to yield a ring expanded ketone.
Wolff Rearrangement
- The Wolff rearrangement is similar to the Tiffeneau-Demjanov rearrangement because of the key role of a diazo intermediate. The most common variation involves reaction of a ketone with a diazo compound. As shown below, treating cyclobutanone with diazomethane yields a ring expansion reaction to form cyclopentanone via a mechanistic pathway that should look familiar.
- Wolff rearrangements are also useful for ring contraction reactions. As shown below, treating an alpha diazoketone with heat or light in methanol promotes the rearrangement to yield the contracted ester product. We will discuss the mechanism for this reaction in the next problem. Forming the starting alpha diazoketone involves a diazo transfer reaction with a ketone. We will generally not worry about this step and instead start with the alpha diazo ketone.


Curtius Rearrangement
- The Curtius rearrangement generally involves the conversion of a carboxylic acid into an amine with the loss of one carbon. It is similar to the Wolff rearrangement but in place of diazomethane, this reaction uses an azide nucleophile. A generic example, shown below, involves generation of an acid chloride upon treatment of the carboxylic acid with thionyl chloride followed by reaction with sodium azide to promote the rearrangement then addition of water to generate the amine product. Other products formed include nitrogen gas and carbon dioxide.
- As shown below in the full mechanism, the key intermediate is an isocyanate. This forms upon rearrangement of the azide intermediate resulting in loss of nitrogen gas and formation of a new R-N bond. Addition of water to the isocyanate generates a carbamic acid that loses carbon dioxide under the reaction conditions to complete the reaction and form the product amine. Other nucleophiles can be added to the isocyanate intermediate to yield different products including a substituted amide, as shown in the problem below.


Baeyer-Villager Rearrangement
- The Baeyer-Villager rearrangement is the reaction of a peracid with an aldehyde or ketone to yield a carboxylic acid or ester, respectively. There are two possible products for ketones with selectivity generally favoring migration of the larger group. The most common peracid used synthetically is meta-chloroperoxybenzoic acid (MCPBA) which also reacts with alkenes to form epoxides. Beware of this dual reactivity when planning syntheses.
- The Baeyer-Villager rearrangement mechanism is shown below. The peracid adds to the aldehyde or ketone to produce a tetrahedral intermediate. Reforming the carbonyl promotes the rearrangement with H migrating exclusively in the aldehyde substrate to yield the carboxylic acid product. A similar intermediate is formed in the ketone reaction. In this case, the larger alkyl group migrates (related to which group can better stabilize a partial carbocation on the carbonyl carbon in the transition state) to yield the ester product.

Beckmann Rearrangement
- The Beckmann rearrangement converts ketones into amides and is the nitrogen equivalent of the Baeyer-Villager reaction with ketones. The rearrangement is relatively straightforward for reactions of symmetrical ketones, as shown in the example below.
- Treatment of the ketone with hydroxylamine yields an oxime that rearranges upon exposure to sulfuric acid to yield a nitrilium ion. Addition of water completes the mechanism to yield the product amide after tautomerization. The details are shown in the scheme below.
- What about reaction of an unsymmetrical ketone? The structure of the oxime leads directly to the rearrangement outcome. Sterically, the OH in the oxime is oriented away from the larger ketone group. This results in the major or only product resulting from rearrangement of the larger ketone substituent.



- The Beckmann rearrangement isn't always this straightforward, as the following experiment illustrates. Beginning with a mixture of the two oximes, they are treated with tosyl chloride to yield the corresponding tosylates. Heating the tosylates yields four products. Products 1 and 2 are the standard Beckmann rearrangement products. How did products 3 and 4 form? Clearly, something strange is happening here.

- To explain how all four of these products formed simultaneously, we must consider a fragmentation reaction as part of the mechanism. This results when a highly stabilized carbocation intermediate can form which is definitely the case for this substrate. Fragmentation of the tosylates yields tertiary carbocations A and C. These carbocations can combine with either of the nitriles (B or D). So, these four recombination possibilities yield the observed products. This experiment highlights another mechanistic possibility for the Beckmann rearrangement and provides a preview of the mechanism we will focus on in our next section (fragmentations!).

Example 6: The molecule below can be used to synthesize substituted azulenes. Our goal in this problem is to propose a three-step synthesis of this bicyclic cyclopentenone. One step involves dichloroketene while another is a ring expansion reaction.

Ans: The key step in this synthesis is a Wolff rearrangement. The synthesis begins with a ketene [2+2] cycloaddition between cycloheptatriene and dichloroketene. The resulting bicyclic ketone reacts with diazomethane and then undergoes the Wolff rearrangement. This results in a ring expansion to yield the 5,7-bicycle. The final step in the synthesis is an E2 elimination of HCl promoted by triethylamine.

FAQs on Rearrangements - Chemistry Optional Notes for UPSC
| 1. What is the Pinacol Rearrangement? |  |
| 2. What is the Payne Rearrangement? | |
| 3. What is the Benzilic Acid Rearrangement? | |
| 4. What is the Tiffeneau-Demjanov Rearrangement? | |
| 5. What is the Wolff Rearrangement? | |

|
Nov 24, 2024 Last updated |

|
Explore Courses for UPSC exam
|
|
Summary
,Exam
,shortcuts and tricks
,video lectures
,Sample Paper
,Extra Questions
,Rearrangements | Chemistry Optional Notes for UPSC
,Rearrangements | Chemistry Optional Notes for UPSC
,Important questions
,MCQs
,Previous Year Questions with Solutions
,past year papers
,mock tests for examination
,study material
,Objective type Questions
,Free
,Rearrangements | Chemistry Optional Notes for UPSC
,practice quizzes
,ppt
,Semester Notes
,Viva Questions
;






Rearrangements Free PDF Download
Importance of Rearrangements
Rearrangements Notes
Rearrangements UPSC Questions
Study Rearrangements on the App
|
© EduRev
|
Education Revolution
|



Follow Us
|




