Organometallic Ligands | Chemistry Optional Notes for UPSC PDF Download
| Table of contents |
|
| Introduction |
|
| Carbon Monoxide |
|
| Metal Hydrides |
|
| Phosphines |
|
| σ Complexes |
|
| Metal Alkyls |
|
| Carbenes |
|
| π Systems |
|
| Odd Numbered π Systems |
|
Introduction
- There are some classes of ligands and modes of bonding that are important and in some cases unique to organometallic complexes and reactions. Before discussing reactions of organometallic complexes we will start with an overview of ligands common to organometallic complexes.
- In organometallic reactions, ligands can be either spectators, not chemically involved or changed during the reaction, or actors, chemically changed during the reaction. Similar to enzymes, a substrate will often bond to an organometallic catalyst as a ligands undergo a chemical transformation, and leave as a product.
Carbon Monoxide
- Carbon monoxide is a neutral strong field ligand that we have discussed previously. We’ve seen that there are two bonding interactions at play in the metal carbonyl bond: a ligand-to-metal n → dσ interaction (σ donation) and a metal-to-ligand dπ → π* interaction (π accepting). The latter interaction is called backbonding because the metal donates electron density back to the ligand.
 Figure 11.1.1: Orbital interactions in M=C=O.
Figure 11.1.1: Orbital interactions in M=C=O. - CO is a fair σ-donor (or σ-base) and a good π-acceptor (or π-acid). The properties of ligated CO depend profoundly upon the identity of the metal center. More specifically, the electronic properties of the metal center dictate the importance of backbonding in metal carbonyl complexes. Most bluntly, more electron-rich metal centers are better at backbonding to CO. Why is it important to ascertain the strength of backbonding?
 Figure 11.1.1: Orbital interactions in M=C=O.
Figure 11.1.1: Orbital interactions in M=C=O.Backbonding and CO vibrational frequency
- Infrared spectroscopy has famously been used to empirically support the idea of backbonding. The table below arranges some metal carbonyl complexes in “periodic” order and provides the frequency corresponding to the C=O stretching mode. Notice that without exception, every complexed CO has a stretching frequency lower than that of free CO. Backbonding is to blame! When a metal donates electrons into a π* antibonding MO on CO, the C-O bond order is lowered and the bond strength is weakened. According to Hooke's law, assuming all other things are equal, when the force constant (bond strength) decreases the vibrational frequency also decreases. Complexes with the strongest metal-CO backbonding will have the lowest frequency CO bond vibration(s).Figure 11.1.2: CO stretching frequencies in metal-carbonyl complexes.
- The figure above depicts a clear increase in frequency (an increase in C–O bond order) as we move left to right across the periodic table. This finding may seem odd if we consider that the number of d electrons in the neutral metal increases as we move left to right. Shouldn’t metal centers with more d electrons be better at backbonding (and more “electron rich”)? What’s going on here? Recall the periodic trend in orbital energy. As we move left to right, the d orbital energies decrease and the energies of the dπ and π* orbitals separate. As a result, the backbonding orbital interaction becomes worse (remember that strong orbital interactions require well-matched orbital energies) as we move toward the more electronegative late transition metals. We can draw an analogy to enamines and enols from organic chemistry. The more electronegative oxygen atom in the enol is a worse electron donor than the enamine’s nitrogen atom.Figure 11.1.3: The importance of backbonding depends on the electronegativity of the metal and its electron density.
- Of course, the contribution of other ligands on the metal center to backbonding cannot be forgotten, either. Logically, electron-donating ligands will tend to make the backbond stronger (they make the metal a better electron donor), while electron-withdrawing ligands will worsen backbonding. Adding electron-rich phosphine ligands to a metal center, for instance, decreases the CO stretching frequency due to improved backbonding.
 Figure 11.1.2: CO stretching frequencies in metal-carbonyl complexes.
Figure 11.1.2: CO stretching frequencies in metal-carbonyl complexes. Figure 11.1.3: The importance of backbonding depends on the electronegativity of the metal and its electron density.
Figure 11.1.3: The importance of backbonding depends on the electronegativity of the metal and its electron density. CO as a bridging ligand
- Carbonyl ligands are famously able to bridge multiple metal centers. Bonding in bridged carbonyl complexes may be either “traditional” or delocalized, depending on the structure of the complex and the bridging mode.
- The variety of bridging modes stems from the different electron donors and acceptors present on the CO ligand (and the possibility of delocalized bonding). Known bridging modes are shown in the figure below.Figure 11.1.4: Building bridges with carbonyl ligands.
 Figure 11.1.4: Building bridges with carbonyl ligands.
Figure 11.1.4: Building bridges with carbonyl ligands.Metal Hydrides
- Metal hydrides occupy an important place in transition metal organometallic chemistry as the M−H bonds can undergo insertion reactions with a variety of unsaturated organic substrates yielding numerous organometallic compounds with M−C bonds. Not only the metal hydrides are needed as synthetic reagents for preparing the transition metal organometallic compounds but they also are required for important hydride insertion steps in many catalytic processes.
- The first transition metal hydride compound was reported by W. Heiber in 1931 when he synthesized Fe(CO)4H2. Though he claimed that the Fe(CO)4H2 contained Fe−H bond, it was not accepted until 1950s, when the concept of normal covalent M−H bond was widely recognized.

Spectroscopic identification
- The metal hydride moieties are easily detectable in 1H NMR as they appear upfield of TMS in the region between 0 to -60 ppm, where no other H resonances appear. The hydride moieties usually couple with metal centers possessing nuclear spins. Similarly, the hydride moieties also couple with the adjacent metal bound phosphine ligands, if present in the complex, exhibiting characteristic cis (J = 15 − 30 Hz) and trans (J = 90 − 150 Hz) coupling constants.
- In the IR spectroscopy, the M−H frequencies appear between (1500 − 2200) cm−1 but their intensities are mostly weak. Crystallographic detection of metal hydride moiety is difficult as hydrogen atoms in general are poor scatterer of X−rays. Located adjacent to a metal atom in a M−H bond, the detection of hydrogen atom thus becomes challenging and as a consequence the X−ray crystallographic method systematically underestimates the M−H internuclear distance by ~ 0.1 Å.
- However, better data could be obtained by performing the X−ray diffraction studies at a low temperature in which the thermal motion of the atoms are significantly reduced. In light of these facts, the neutron diffraction becomes a powerful method for detection of the metal hydride moieties as hydrogen scatters neutrons more effectively and hence the M−H bond distances can be measured more accurately. A limitation of neutron diffraction method is that large sized crystals are required for the study.
Bridging hydrides
- Bridging hydrides are an intriguing class of ligands. A question to ponder: how can a ligand associated with only two electrons possibly bridge two metal centers? How can two electrons hold three atoms together? Enter the magic of three-center, two-electron bonding.
- We can envision the M–H sigma bond as an electron donor itself! With this in mind, we can imagine that hydrides are able to bind end-on to one metal and side-on to another. Consistent with the idea that bridging is the result of “end-on + side-on” bonding, bond angles of bridging hydrides are never 180°.Figure 11.1.5: Resonance forms of bridging hydrides, with an example.
 Figure 11.1.5: Resonance forms of bridging hydrides, with an example.
Figure 11.1.5: Resonance forms of bridging hydrides, with an example.Phosphines
- Phosphines are most notable for their remarkable electronic and steric tunability and their “innocence”—they tend to avoid participating directly in reactions, but have the ability to profoundly modulate the electronic properties of the metal center to which they’re bound.
- Furthermore, because the energy barrier to inversion is quite high, “chiral-at-phosphorus” ligands can be isolated in enantioenriched form and introduced to metal centers, bringing asymmetry just about as close to the metal as it can get in chiral complexes.
- The one stable isotope of P (31P) is NMR active so 31P NMR is often used to cahrecterize complexes and reactions involving phosphine ligands . Soft phosphines match up very well with the soft low-valent transition metals. Electron-poor phosphines are even good π-acids.
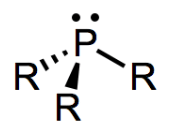
Bonding
- Like CO, phosphines are neutral σ donors and π acceptors that formally contribute two electrons to the metal center. Unlike CO, most phosphines are not small enough for more than four to bond to a single metal center (and for large R, the number is even smaller). Steric hindrance becomes a problem when five or more PR3 ligands try to make their way into the space around the metal. Bridging by phosphines is extremely rare, but ligands containing multiple phosphine donors such as diphenylphosphinoethane metal center are common.
- For entropic reasons, chelating ligands like these bind to a single metal center at multiple points if possible, instead of attaching to two different metal centers (the aptly named chelate effect). An important characteristic of chelating phosphines is bite angle, defined as the predominant P–M–P angle in known complexes of the ligand. When the preferred bite angle of the ligand doesn't match the ideal bond angles of the geometry (i.e. 90° for octahedral) that introduces strain, and potentially reactivity into the complex.
- The predominant orbital interaction contributing to phosphine binding is the one we expect, a lone pair on phosphorus donating to an empty metal d orbital. The electronic nature of the R groups influences the electron-donating ability of the phosphorus atom. For instance, alkylphosphines, which possess P–Csp3 bonds, tend to be better electron donors than arylphosphines, which possess P–Csp2 bonds.
- The rationale here is the greater electronegativity of the sp2 hybrid orbital versus the sp3 hybrid, which causes the phosphorus atom to hold more tightly to its lone pair when bound to an sp2 carbon. The same idea applies when electron-withdrawing and -donating groups are incorporated into R: the electron density on P is low when R contains electron-withdrawing groups and high when R contains electron-donating groups.Figure 11.1.6: As we add electronegative R groups, the phosphorus atom (and the metal to which it's bound) become more electron poor.
- Like CO, phosphines participate in backbonding to a certain degree; however, the phenomenon here is of a fundamentally different nature than CO backbonding. For one thing, phosphines lack a π* orbital. In the days of yore, chemists attributed backbonding in phosphine complexes to an interaction between a metallic dπ orbital and an empty 3d orbital on phosphorus. However, this idea has elegantly been proven bogus, and a much more organic-friendly explanation has taken its place (no d orbitals on P required). In an illuminating series of experiments, M–P and P–R bond lengths were measured via crystallography for several redox pairs of complexes.
- Oxidation decreases the ability of the metal to backbond, because it removes electron density from the metal. This explains the increases in M–P bond length—just imagine a decrease the M–P bond order due to decreased backbonding. And the decrease in P–R bond length? It’s important to see that invoking only the phosphorus 3d orbitals would not explain changes in the P–R bond lengths, as the 3d atomic orbitals are most definitely localized on phosphorus.
- Instead, we must invoke the participation of σ*P–R orbitals in phosphine backbonding to account for the P–R length decreases. The figure below depicts one of the interactions involved in M–P backbonding, a dπ → σ* interaction (an orthogonal dπ → σ* interaction also plays a role). As with CO, a resonance structure depicting an M=P double bond is a useful heuristic! Naturally, R groups that are better able to stabilize negative charge—that is, electron-withdrawing groups—facilitate backbonding in phosphines. Electron-rich metals help too.
 Figure 11.1.6: As we add electronegative R groups, the phosphorus atom (and the metal to which it's bound) become more electron poor.
Figure 11.1.6: As we add electronegative R groups, the phosphorus atom (and the metal to which it's bound) become more electron poor.

Cone Angle
- The steric and electronic properties of phosphines vary enormously. Tolman devised some intriguing parameters that characterize the steric and electronic properties of this class of ligands. To address sterics, he developed the idea of cone angle—the apex angle of a cone formed by a point 2.28 Å from the phosphorus atom (an idealized M–P bond length), and the outermost edges of atoms in the R groups, when the R groups are folded back as much as possible. Wider cone angles, Tolman reasoned, indicate greater steric congestion around the phosphorus atom. To address electronics, Tolman used the CO stretching frequency (νCO) of mixed phosphine-carbonyl complexes. Specifically, he used Ni(CO)3L complexes, where L is a tertiary phosphine, as his standard.Figure 11.1.9: Illustration of the cone angle in phosphines.
- Tolman’s logic went as follows: more strongly electron-donating phosphines are associated with more electron-rich metals, which are better at CO backbonding (due fundamentally to higher orbital energies). Better CO backbonding corresponds to a lower νCO due to decreased C–O bond order. Thus, better donor ligands should be associated with lower νCO values (and vice versa for electron-withdrawing ligands). Was he correct? Exhibit A…Notice the trifluorophosphine stuck in the “very small, very withdrawing” corner, and its utter opposite, the gargantuan tri(tert-butyl)phosphine in the “extremely bulky, very donating” corner.
 Figure 11.1.9: Illustration of the cone angle in phosphines.
Figure 11.1.9: Illustration of the cone angle in phosphines. Notice the trifluorophosphine stuck in the “very small, very withdrawing” corner, and its utter opposite, the gargantuan tri(tert-butyl)phosphine in the “extremely bulky, very donating” corner.
Notice the trifluorophosphine stuck in the “very small, very withdrawing” corner, and its utter opposite, the gargantuan tri(tert-butyl)phosphine in the “extremely bulky, very donating” corner.σ Complexes
General Properties
- Previously our discussion of metal-ligand σ bonding was limited to ligand lone pairs acting as donors. Ligands can also use a filled σ bonding molecular orbital to donate to a metal as shown in Figure 11.1.10. This binding mode results in side-on η2 bonding. If the backbonding interaction is too strong the single L-L ligands can become two L- ligands by breaking the L-L sigma bond.Figure 11.1.10: Sigma bonding (left) and pi backbonding (right) orbital interaction in metal σ complexes
- The first thing to realize about σ complexes is that they are highly sensitive to steric bulk. Any old σ bond won’t do; hydrogen at one end of the binding bond or the other (or both) is necessary. The best studied σ complexes involve dihydrogen (H2), so let’s start there.
 Figure 11.1.10: Sigma bonding (left) and pi backbonding (right) orbital interaction in metal σ complexes
Figure 11.1.10: Sigma bonding (left) and pi backbonding (right) orbital interaction in metal σ complexes Dihydrogen complexes
Mildly backbonding metals may bind dihydrogen “side on.” Like side-on binding in π complexes, there are two important orbital interactions at play here: σH–H→dσ and dπ→σ*H–H. Dihydrogen complexes can “tautomerize” to (H)2 isomers through oxidative addition of the H–H bond to the metal. We should expect more electron-rich metal centers to favor the X2 isomer, since these should donate more strongly into the σ*H–H orbital.
This idea was masterfully demonstrated in a study by Morris, in which he showed that H2 complexes of π-basic metal centers show all the signs of dihydride complexes, rather than hydrogen complexes. More generally, metal centers in σ complexes need a good balance of π basicity and σ acidity (I like to call this the “Goldilocks effect”). Because of the need for balance, σ complexes are most common for centrally located metals (groups 6-9).

Metal Alkyls

- Alkyl or aryl transition metal compounds have M-C single bonds. In spite of many attempts over most of the course of chemical history, their isolation was unsuccessful and it was long considered that all M-C bonds were essentially unstable. Stable alkyl complexes began to be prepared gradually only from the 1950s. Cp2ZrCl(Pr),WMe6, CpFeMe(CO)2, CoMe(py)(dmg)2, (dmg = dimethylglyoximato), IrCl(X)(Et)(CO)(PPh3)2, NiEt2(bipy), PtCl(Et)(PEt3)2 are some representative compounds.
- Among various synthetic processes so far developed, the reactions of compounds containing M-halogen bonds with main-group metal-alkyl compounds, such as a Grignard reagent or an organolithium compound, are common synthetic routes. Especially vitamin B12, of which Dorothy Hodgkin (1964 Nobel Prize) determined the structure, is known to have a very stable Co-C bond. Metal alkyl compounds which have only alkyl ligand, such as WMe6, are called homoleptic alkyls.
- It is gradually accepted that a major cause of the instability of alkyl complexes is the low activation energy of their decomposition rather than a low M-C bond energy. The most general decomposition path is β elimination. Namely, the bonding interaction of a hydrocarbon ligand with the central transition metal tends to result in the formation of a metal hydride and an olefin.
- Such an interaction is called an agostic interaction. Although an alkyl and an aryl ligand are 1-electron ligands, they are regarded as anions when the oxidation number of the metal is counted. The hydride ligand, H, resembles the alkyl ligand in this aspect.
Carbenes
- In this section we’ll investigate two classes of carbenes, which are characterized by a metal-carbon double bond. Fischer carbenes and Shrock carbenes are usually actor ligands, but they may be either nucleophilic or electrophilic, depending on the nature of the R groups and metal.
- In addition, these ligands present some interesting synthetic problems: because free carbenes are quite unstable, ligand substitution reactons can't be used for metal carbene synthesis.

Fisher vs Schrock carbenes
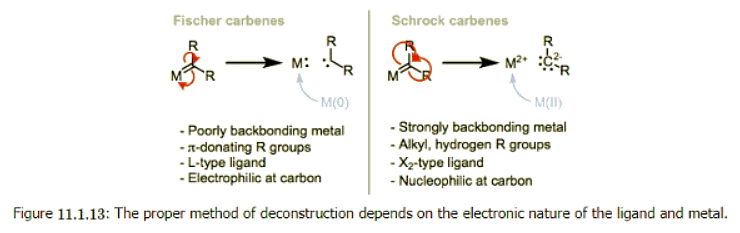
- Metal carbenes all possess a metal-carbon double bond. What’s interesting for us about this double bond is that there are multiple ways to deconstruct it to determine the metal’s oxidation state and number of d electrons. We could give one pair of electrons to the metal center and one to the ligand. This procedure nicely illustrates why compounds containing M=C bonds are called “metal carbenoids”—the deconstructed ligand is a neutral carbenoid. Alternatively, we could give both pairs of electrons to the ligand and think of it as a dianionic ligand. The appropriate procedure depends on the ligand’s substituents and the electronic nature of the metal. The figure below summarizes the two deconstruction procedures.
- When the metal possesses π-acidic ligands and the R groups are π-basic, the complex is best described as an L-type Fischer carbene and the oxidation state of the metal is unaffected by the carbene ligand. When the ligands are “neutral” (R = H, alkyl) and the metal is a good backbonder—that is, in the absence of π-acidic ligands and electronegative late metals—the complex is best described as an anionic X2-type Schrock carbene. Notice that the oxidation state of the metal depends on our deconstruction method. In organometallic complexes it isn't uncommon for the oxidation state of the metal and ligand to be ambiguous.
- Deconstruction reveals the typical behavior of the methylene carbon in each class of complex. The methylene carbon of Schrock carbenes, on which electron density is piled through backbonding, is nucleophilic. On the other hand, the methylene carbon of Fischer carbons is electrophilic, because backbonding is weak and does not compensate for σ-donation from the ligand to the metal. To spot a Fischer carbene, be on the lookout for reasonable zwitterionic resonance structures like the one at right below.
- The clever reader may notice that we haven’t mentioned π-acidic R groups, such as carbonyls. Complexes of this type are best described as Fischer carbenes as well, as the ligand is still electrophilic. However, complexes of this type are difficult to handle and crazy reactive (see below) without a π-basic substituent to hold them in check. Take care when diagnosing the behavior of metal carbenes. In these complexes, there is often a subtle interplay between the R groups on the carbene and other ligands on the metal. In practice, many carbenes are intermediate between the Fischer and Schrock ideals.
 The clever reader may notice that we haven’t mentioned π-acidic R groups, such as carbonyls. Complexes of this type are best described as Fischer carbenes as well, as the ligand is still electrophilic. However, complexes of this type are difficult to handle and crazy reactive (see below) without a π-basic substituent to hold them in check. Take care when diagnosing the behavior of metal carbenes. In these complexes, there is often a subtle interplay between the R groups on the carbene and other ligands on the metal. In practice, many carbenes are intermediate between the Fischer and Schrock ideals.
The clever reader may notice that we haven’t mentioned π-acidic R groups, such as carbonyls. Complexes of this type are best described as Fischer carbenes as well, as the ligand is still electrophilic. However, complexes of this type are difficult to handle and crazy reactive (see below) without a π-basic substituent to hold them in check. Take care when diagnosing the behavior of metal carbenes. In these complexes, there is often a subtle interplay between the R groups on the carbene and other ligands on the metal. In practice, many carbenes are intermediate between the Fischer and Schrock ideals.π Systems
- In contrast to neutral spectator ligands, π systems most often play an important role in the reactivity of the organometallic complexes of which they are a part (since they act in reactions, they’re called “actors”). π systems do useful chemistry, not just with the metal center, but also with other ligands and external reagents.
- Thus, in addition to thinking about how π systems affect the steric and electronic properties of the metal center, we need to start considering the metal’s effect on the ligand and how we might expect the ligand to behave as an active participant in reactions.
- To the extent that structure determines reactivity—a commonly repeated, and extremely powerful maxim in organic chemistry—we can think about possibilities for chemical change without knowing the elementary steps of organometallic chemistry in detail yet.
General Properties
- Similar to σ complexes, the π bonding orbitals of alkenes, alkynes, carbonyls, and other unsaturated compounds may overlap with dσ orbitals on metal centers. This is the classic ligand HOMO → metal LUMO interaction that we’ve seen many times. Because of this electron donation from the π system to the metal center, coordinated π systems often act electrophilic, even if the starting alkene was nucleophilic.
- The π → dσ orbital interaction is central to the structure and reactivity of π-system complexes. π systems are also often subject to important backbonding interactions. We’ll focus on alkenes here, but these same ideas apply to carbonyls, alkynes, and other unsaturated ligands bound through their π clouds. For alkene ligands, the relative importance of “normal” bonding and backbonding is nicely captured by the relative importance of the two resonance structures in Figure 11.1.14.
- Complexes of weakly backbonding metals, such as the electronegative late metals, are best represented by the traditional dative resonance structure 1. But complexes of strong backbonders, such as electropositive Ti(II), are often best drawn in the metallacyclopropane form 2. Bond lengths and angles in the alkene change substantially upon coordination to a strongly backbonding metal. We see an elongation of the C=C bond (consistent with decreased bond order) and some pyramidalization of the alkene carbons (consistent with a change in hybridization from sp2 to sp3). A complete orbital picture of sigma bonding and pi backbonding in alkenes is shown in Figure 11.1.15.
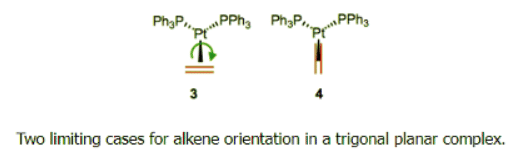
- Here’s an interesting question with stereochemical implications: what is the orientation of the alkene relative to the other ligands? From what we’ve discussed so far, we can surmise that one face of the alkene must point toward the metal center. Put differently, the bonding axis must be normal to the plane of the alkene. However, this restriction says nothing about rotation about the bonding axis, which spins the alkene ligand like a pinwheel. Is a particular orientation preferred, or can we think about the alkene as a circular smudge over time? The figure below depicts two possible orientations of the alkene ligand in a trigonal planar complex. Other orientations make less sense because they would involve inefficient orbital overlap with the metal’s orthogonal d orbitals. Which one is favored?
- First of all, we need to notice that these two complexes are diastereomeric. They have different energies as a result, so one must be favored over the other. Steric considerations suggest that complex 4 ought to be more stable (in most complexes, steric factors dictate alkene orientation). To dig a little deeper, let’s consider any electronic factors that may influence the preferred geometry. We’ve already seen that electronic factors can overcome steric considerations when it comes to complex geometry. To begin, we need to consider the crystal field orbitals of the complex as a whole. Verify on your own that in this d10, Pt(0) complex, the crystal-field HOMOs are the dxy and d-{x^2–y^2} orbitals. Where are these orbitals located in space? In the xy-plane! Only the alkene in 3 can engage in efficient backbonding with the metal center. In cases when the metal is electron rich and/or the alkene is electron poor, complexes like 3 can sometimes be favored in spite of sterics.


Arenes
- Arenes or aromatic ligands are neutral ligands that may serve either as actors or spectators. Arenes commonly bind to metals through more than two atoms, although η2-arene ligands are known. Structurally, most η6-arenes tend to remain planar after binding to metals. Both sigma bonding and pi backbonding are possible for arene ligands; however, arenes are stronger sigma donors than CO and backbonding is less important for these ligands.
- The reactivity of arenes changes dramatically upon metal binding, along lines that we would expect for strongly electron-donating ligands. After coordinating to a transition metal, the arene usually becomes a better electrophile (particularly when the metal is electron poor). Thus, metal coordination can enable otherwise difficult nucleophilic aromatic substitution reactions.
General Properties
- The coordination of an aromatic compound to a metal center through its aromatic π MOs removes electron density from the ring. π → dσ (sigma bonding) and dπ → π* (backbonding) orbital interactions are possible for arene ligands, with the former being much more important, typically. To simplify drawings, you often see chemists draw arenes involving a circle and single central line to represent the π → dσ orbital interaction. Despite the single line these are not simple 2 electron donors. For instance, η6-arenes are best describes as six-electron donors.
- Multiple coordination modes are possible for arene ligands. When all six atoms of a benzene ring are bound to the metal (η6-mode), the ring is flat and C–C bond lengths are slightly longer than those in free benzene. The ring is bent and non-aromatic in η4-mode, so that the four atoms bound to the metal are coplanar while the other π bond is out of the plane. Even η2-arene ligands bound through one double bond are known.
- Coordination of one π bond results in dearomatization and makes η2-benzene behave more like butadiene, and furan act more like a vinyl ether. With naphthalene as ligand, there are multiple η2 isomers that could form; the isomer observed is the one that retains aromaticity in the free portion of the ligand. In fact, this result is general for polycyclic aromatic hydrocarbons: binding maximizes aromaticity in the free portion of the ligand. Arene ligands are usually hydrocarbons, not heterocycles. Why? Aromatic heterocycles, such as pyridine, more commonly bind using their basic lone pairs. That said, a few heterocycles form important π complexes. Thiophene is perhaps the most heavily studied, as the desulfurization of thiophene from fossil fuels is an industrially useful process.
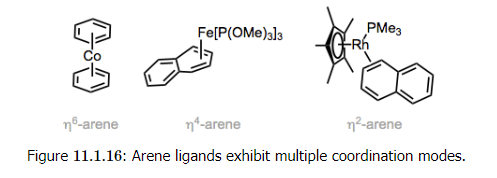
Odd Numbered π Systems
- Odd-numbered π systems—most notably, the allyl and cyclopentadienyl ligands—are formally monoanionic ligands which donate n+1 electrons (i.e. η5 cyclopentadienyl ligand is a 6 electron donor). To illustrate the plurality of equally important resonance structures for this class of ligands, we often just draw a curved line from one end of the π system to the other.
- Yet, even this form is not perfect, as it obscures the possibility that the datively bound atoms may dissociate from the metal center, forming σ-allyl or ring-slipped ligands. What do the odd-numbered π systems really look like, and how do they really behave?

Metal Allyls
- Allyls are often actor ligands, most famously in allylic substitution reactions. The allyl ligand is an interesting beast because it may bind to metals in two ways. When its double bond does not become involved in binding to the metal, allyl is a simple anionic ligand bound covalently through one carbon—basically, a monodentate alkyl! Alternatively, allyl can act as a bidentate LX-type ligand, bound to the metal through all three conjugated atoms. The LX or “trihapto” form can be represented using one of two resonance forms, or (more common) the “aromatic” form seen in the illustration above.Can we use FMO theory to explain the wonky geometry of the allyl ligand?
- The lower half of the figure above illustrates the slightly weird character of the geometry of allyl ligands. In a previous post on even-numbered π systems, we investigated the orientation of the ligand with respect to the metal and came to some logical conclusions by invoking FMO theory and backbonding. A similar treatment of the allyl ligand leads us to similar conclusions: the plane of the allyl ligand should be parallel to the xy-plane of the metal center and normal to the z-axis. In reality, the allyl plane is slightly canted to optimize orbital overlap—but we can see at the right of the figure above that π2–dxy orbital overlap is key. Also note the rotation of the anti hydrogens (anti to the central C–H, that is) toward the metal center to improve orbital overlap.
- Exchange of the syn and anti substituents can occur through σ,π-isomerization followed by bond rotation and formation of the isomerized trihapto form. Notice that the configuration of the stereocenter bearing the methyl group is unaffected by the isomerization! It should be noted that 1,3-disubstituted allyl complexes almost exclusively adopt a syn,syn configuration without danger of isomerization.
 Can we use FMO theory to explain the wonky geometry of the allyl ligand?
Can we use FMO theory to explain the wonky geometry of the allyl ligand?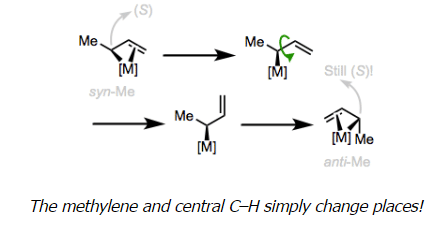
Metal Cyclopentadienyls
- Upon deconstruction, the cyclopentadienyl (Cp) ligand yields the aromatic cyclopentadienyl anion, an L2X-type ligand. Cp is normally an η5-ligand, but η3 (LX) and η1 (X) forms are known in cases where the other ligands on the metal center are tightly bound. η1-Cyclopentadienyl ligands can sometimes be fluxional—the metal has the ability to “jump” from atom to atom.
- Variations on the Cp lignad include Cp* (C5Me5) and the monomethyl version (C5H4Me). A single Cp may bond alongside other ligands (in “half-sandwich” or “piano-stool” complexes), or paired up with a second Cp ligand in metallocenes. The piano-stool and bent metallocene complexes are most interesting for us, since these have potential for open coordination sites—metallocenes tend to be relatively stable and boring.

FAQs on Organometallic Ligands - Chemistry Optional Notes for UPSC
| 1. What is the role of metal alkyls in organometallic chemistry? |  |
| 2. How do metal hydrides contribute to organometallic chemistry? | |
| 3. What are the characteristics of phosphines as organometallic ligands? | |
| 4. How do π systems contribute to organometallic chemistry? | |
| 5. What are σ complexes in organometallic chemistry? | |

|
Dec 18, 2024 Last updated |

|
Explore Courses for UPSC exam
|
|
Important questions
,ppt
,Organometallic Ligands | Chemistry Optional Notes for UPSC
,MCQs
,study material
,Viva Questions
,mock tests for examination
,Previous Year Questions with Solutions
,practice quizzes
,Extra Questions
,past year papers
,Summary
,shortcuts and tricks
,Organometallic Ligands | Chemistry Optional Notes for UPSC
,Organometallic Ligands | Chemistry Optional Notes for UPSC
,video lectures
,Objective type Questions
,Exam
,Free
,Semester Notes
,Sample Paper
;






Organometallic Ligands Free PDF Download
Importance of Organometallic Ligands
Organometallic Ligands Notes
Organometallic Ligands UPSC Questions
Study Organometallic Ligands on the App
|
© EduRev
|
Education Revolution
|



Follow Us
|




